 夜雨聆风
夜雨聆风高熵合金(high-entropy alloys, HEAs)因其丰富的元素组合、可调电子结构和优异稳定性,正在成为电催化材料设计中的重要平台。然而,也正是因为其元素空间极其庞大、元素间协同作用复杂,如何高效筛选出真正高活性的组合,并进一步理解“为什么有效”,一直是该领域的核心难题。
近日,顶刊 National Science Review 发表研究论文 “Unveiling the correlation between high-entropy alloy element systems and electrocatalytic activity”。该工作由中国科学院长春应用化学研究所/中国科学技术大学汪尔康院士、周敏研究员团队,华南理工大学团队,以及日本东北大学李昊杰出教授团队合作完成。研究提出了一套将大语言模型/AI Agent、高通量自动化实验平台与精准理论建模相结合的协同框架,用于揭示高熵合金元素体系与氧还原反应(ORR)活性之间的关联。
从“盲目组合”到“AI 辅助枚举”
高熵合金的优势在于多元素协同,但挑战也来自多元素组合。传统机器学习方法通常依赖结构化数据,适合在已有数据集中做性能预测或优化;但对于高熵合金这样知识分散、文献信息复杂、实验空间巨大的体系,仅靠传统模型很难充分利用文献中的非结构化知识,也难以贯穿“材料设计—实验规划—数据分析—机理解释”的完整流程。
为此,作者构建了面向高熵合金电催化研究的 AI 助手 ChatHEA。研究首先利用 GPT-4o API 对约 17,000 篇 ORR 相关摘要进行文本挖掘,提取 ORR 研究中的元素库;随后进一步从 200 篇精选高熵合金相关文献中抽取领域知识,并经专家审核、清洗与结构化,构建高熵合金电催化知识数据集。在此基础上,作者采用 LoRA 方法对 Llama-3-8B 进行领域微调,形成了专用于 HEA 电催化研究的 ChatHEA。
需要强调的是,ChatHEA 在这项工作中并不是一个简单的“黑箱预测器”,而是一个研究流程中的智能中枢。它的作用包括:根据文献先验和化学直觉枚举高熵合金组合,辅助设计实验方案,组织高通量实验任务,并对获得的数据进行自动化处理与多维关联分析。
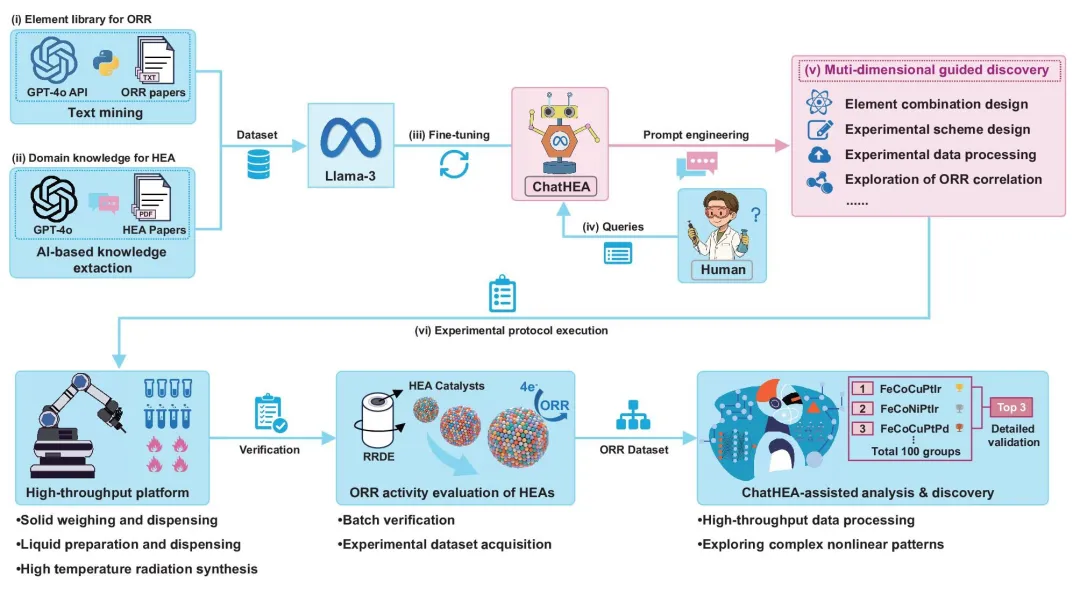
图 1|大语言模型与高通量实验平台的协同框架。图中嵌入的 GPT 和 Llama 标志表示该流程由相应模型驱动,这些标志分别归 OpenAI 和 Meta 所有。双蛇 Python 图标表示使用了 Python,其归 Python Software Foundation 所有。
从图 1 可以看到,这一框架并不是单点式 AI 预测,而是一个较完整的协同流程:首先通过 GPT-4o 对 ORR 文献和 HEA 文献进行知识抽取,随后利用 Llama-3 微调得到 ChatHEA;ChatHEA 再通过自然语言交互,辅助完成元素组合设计、实验方案设计、实验数据处理以及 ORR 相关性探索。最后,高通量平台负责执行合成和测试,形成标准化 ORR 数据集,再由 ChatHEA 进行分析和发现。
100 组高熵合金:高通量合成与 ORR 数据集构建
在 ChatHEA 的辅助下,研究团队设计并枚举了 100 组五元高熵合金组合。这些组合并非随机选择,而是在文献先验、元素性质、前驱体可行性和化学约束下形成的系统性样品库,目的是让不同元素体系之间具有可比性,从而用于发现“元素组合—ORR 活性”的内在规律。
随后,作者利用高通量热辐射合成平台,在 CO₂ 活化碳纳米纤维基底上快速制备了 100 组 HEA 催化剂,并通过旋转环盘电极(RRDE)系统评价其 ORR 性能。整个 ORR 活性测试在 5 个工作日内完成,形成了标准化的 ORR 活性数据集。
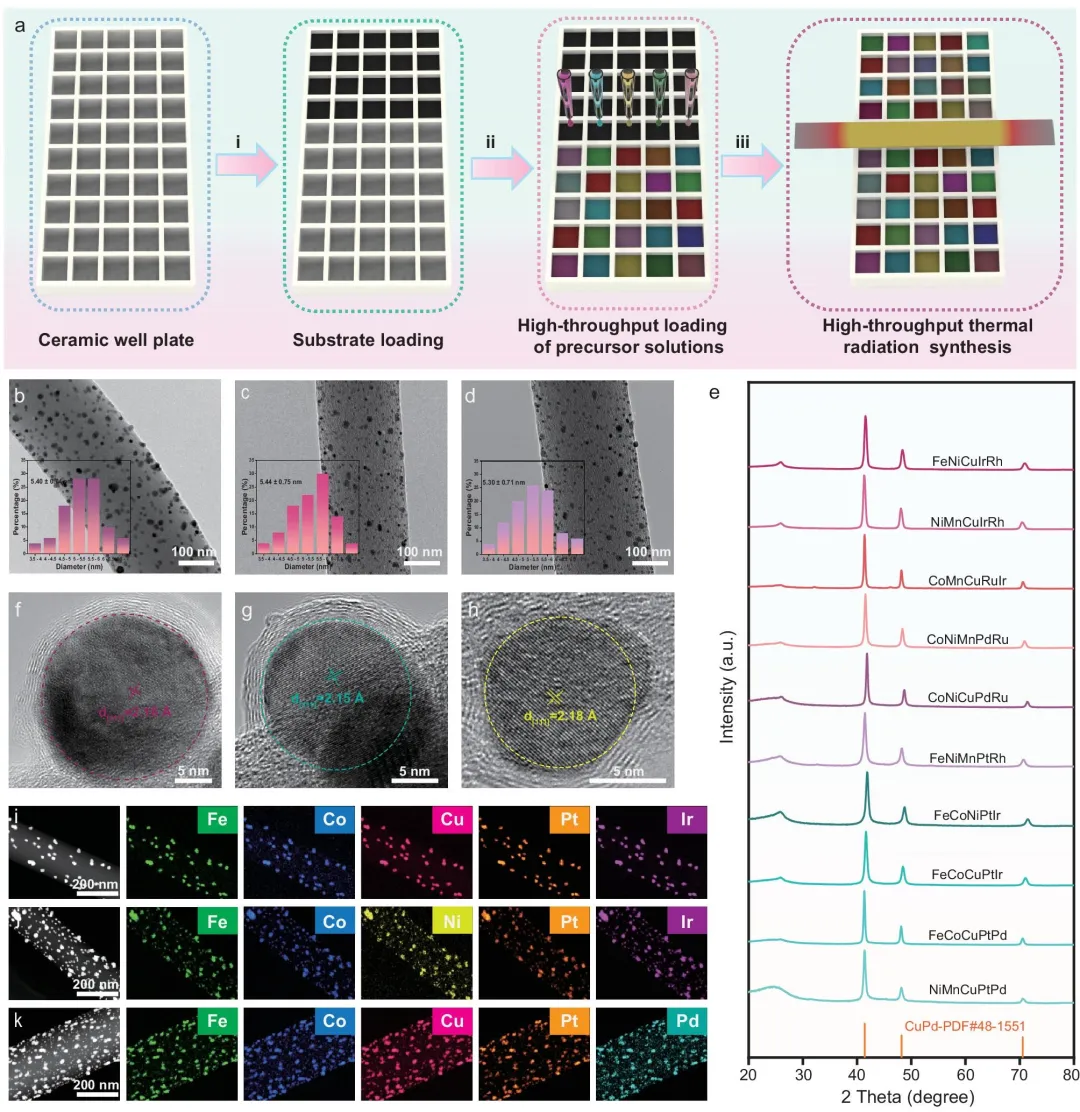
图 2|HEA 催化剂的高通量合成与表征。(a) HEA 催化剂高通量热辐射合成的流程示意图。(b–d) FeCoCuPtIr/FeCoNiPtIr/FeCoCuPtPd@CA-CNFs 的 TEM 图像,插图为 HEA 纳米颗粒的粒径分布直方图。(e) HEAs@CA-CNFs 的 XRD 图谱。(f–h) FeCoCuPtIr/FeCoNiPtIr/FeCoCuPtPd@CA-CNFs 的 HRTEM 图像。(i–k) 相应的 HAADF-STEM 图像及 EDS 元素映射图。
材料表征结果显示,不同元素组合的 HEA 纳米颗粒具有相近的负载密度和粒径分布,XRD 表明其主要呈面心立方结构,HRTEM 和 HAADF-STEM-EDS 映射进一步确认了多元素在纳米颗粒中的均匀分布。这说明高通量制备得到的样品具有较好的结构一致性,为后续比较不同元素组合的 ORR 活性提供了可靠基础。
ChatHEA 揭示关键元素协同:Fe–Co–Cu / Fe–Co–Ni 与 Pt–Ir / Pt–Pd
在获得标准化 ORR 活性数据集后,ChatHEA 对 100 组 HEA 催化剂的半波电位 E₁/₂ 进行了相关性和特征重要性分析。
在线性模型层面,Pearson 相关系数、Lasso 和 Ridge 回归均显示,非贵金属元素体系中的 Fe–Co–Cu、Fe–Co–Ni、Fe–Ni–Cu,以及贵金属元素体系中的 Pt–Ir、Pt–Pd,与 ORR 活性呈现显著正相关。
有意思的是,当分析维度降到单元素水平时,只有 Pt 和 Fe 显示出明显正相关,而 Co、Ni、Cu 的单独贡献并不突出。这说明高熵合金的 ORR 活性并不是由某一个元素简单线性决定,而是来自多元素之间的协同耦合。
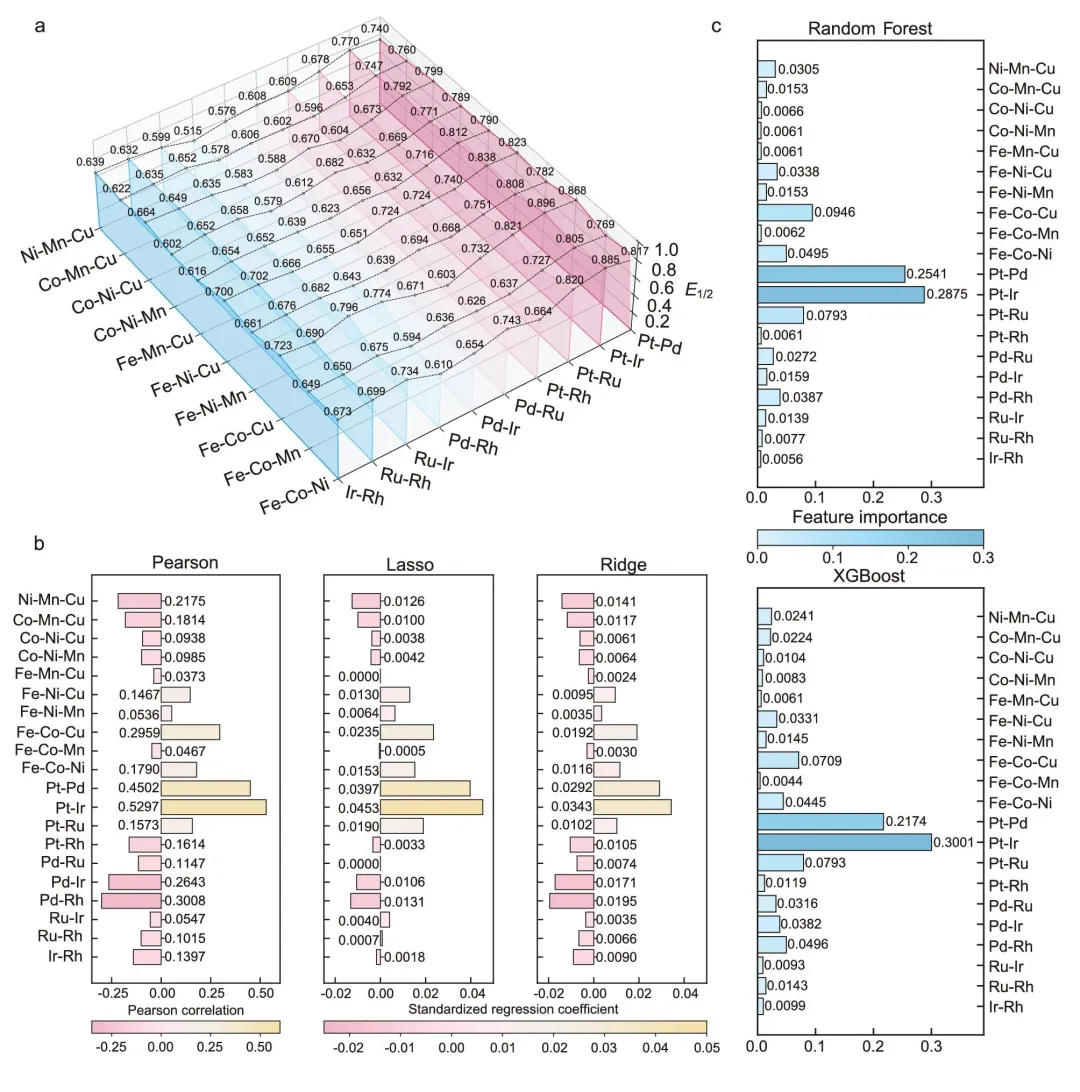
图 3|100 组 HEA 组合的 ORR 活性数据集及相关性/特征重要性分析。(a) 100 组 HEA 组合的半波电位 E₁/₂ 数据集。(b) 基于 Pearson 相关性、Lasso 和 Ridge 回归模型,分析非贵金属/贵金属元素体系与 E₁/₂ 之间的相关性。(c) 基于 Random Forest 和 XGBoost 模型,分析非贵金属/贵金属元素体系与 E₁/₂ 之间的特征重要性。
进一步地,Random Forest 和 XGBoost 等非线性模型也给出了类似结论:Fe–Co–Cu、Fe–Co–Ni、Fe–Ni–Cu、Pt–Ir 和 Pt–Pd 是影响 ORR 活性的关键元素体系。不同模型之间的一致性,说明该结论具有较好的稳健性,也为后续筛选高性能组合提供了明确方向。
最优组合 FeCoCuPtIr:活性、稳定性与燃料电池验证
基于 ChatHEA 的分析,研究进一步筛选出三组高性能 HEA 组合:FeCoCuPtIr、FeCoNiPtIr 和 FeCoCuPtPd,并进行了详细实验验证。
其中,FeCoCuPtIr 表现最优。其 ORR 半波电位达到 0.894 V vs. RHE,Tafel 斜率为 65.8 mV dec⁻¹,均优于商业 Pt/C。更重要的是,在 0.85 V vs. RHE 下,三种 HEA 催化剂的质量活性分别为 1.15、0.84 和 0.66 A mg⁻¹,对应为 Pt/C 的 10.5、7.6 和 6.0 倍。RRDE 分析显示,这些催化剂主要遵循 4 电子 ORR 路径,H₂O₂ 产率较低。
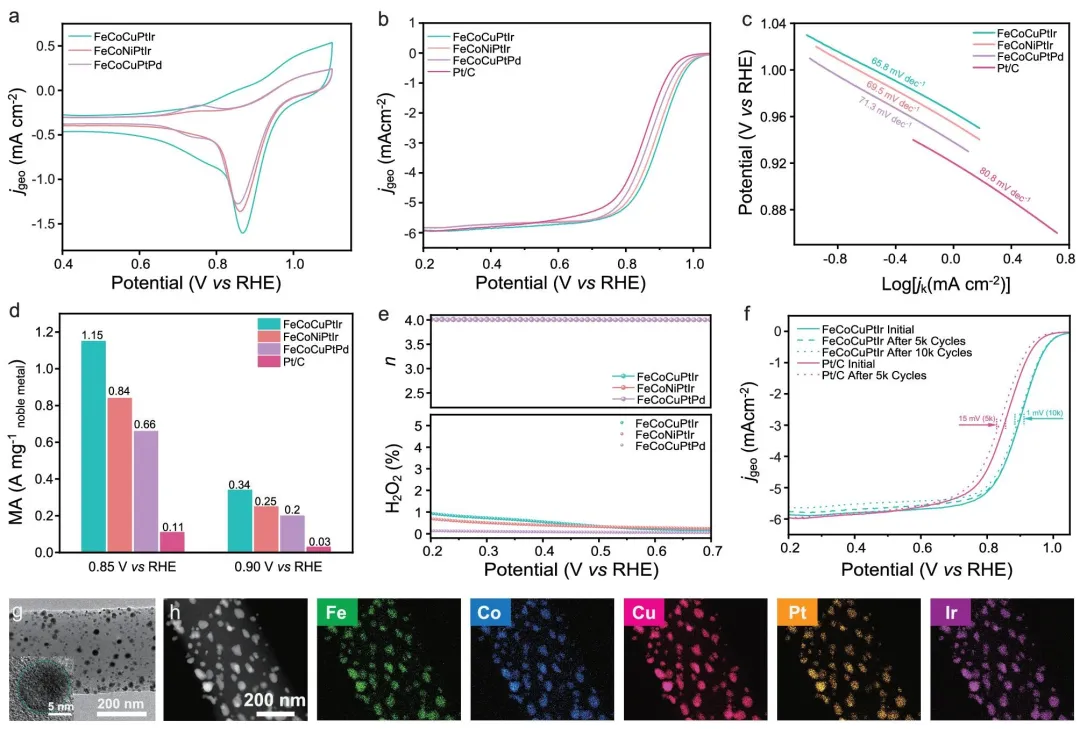
图 4|高性能 HEA 组合的 ORR 性能评价。(a) 三种 HEA 催化剂的循环伏安曲线。(b) 三种 HEA 催化剂和 Pt/C 在 1600 r/min 下的线性扫描伏安曲线。(c) 三种 HEA 催化剂和 Pt/C 的 Tafel 曲线。(d) 三种 HEA 催化剂和 Pt/C 的质量活性比较。(e) 三种 HEA 催化剂的电子转移数和 H₂O₂ 产率。(f) FeCoCuPtIr 和 Pt/C 的加速衰减测试比较。(g) 加速衰减测试后 FeCoCuPtIr@CA-CNFs 的 TEM 图像,插图为单个颗粒的 HRTEM 图像。(h) HAADF-STEM 图像及相应的 EDS 元素映射图。
稳定性方面,FeCoCuPtIr 在 5000 圈加速衰减测试后性能几乎没有明显变化,而 Pt/C 的半波电位下降约 15 mV。测试后的形貌和元素分布表明,FeCoCuPtIr 仍能保持纳米颗粒结构,没有明显团聚或相分离,体现出高熵效应带来的结构稳定性。
为了进一步验证实际应用潜力,作者还将 FeCoCuPtIr 用于质子交换膜燃料电池(PEMFC)测试。在 H₂–air 条件下,FeCoCuPtIr 阴极实现了 0.789 W cm⁻² 的峰值功率密度,高于商业 Pt/C 的 0.724 W cm⁻²。在 0.9 V_{iR-free} 下,其质量活性达到 0.806 A mg_{noble metal}⁻¹,约为 DOE 2025 活性目标的 1.83 倍,显示出良好的器件级应用潜力。
精准理论建模:为什么 FeCoCuPtIr 更好?
为了理解高活性的来源,研究进一步结合 DFT 计算与 pH 依赖微观动力学模型进行机理分析。
作者构建了 FeCoCuPtIr、FeCoNiPtIr 和 FeCoCuPtPd 的 HEA-(111) 表面模型,并考察多个代表性吸附位点的 O* 吸附能。结果表明,这些高活性体系中存在接近 ORR 火山图最优区域的吸附位点。差分电荷密度分析显示,O* 吸附过程中界面电荷转移适中,既不会过强导致 O* 过度稳定,也不会过弱而不利于反应物活化。
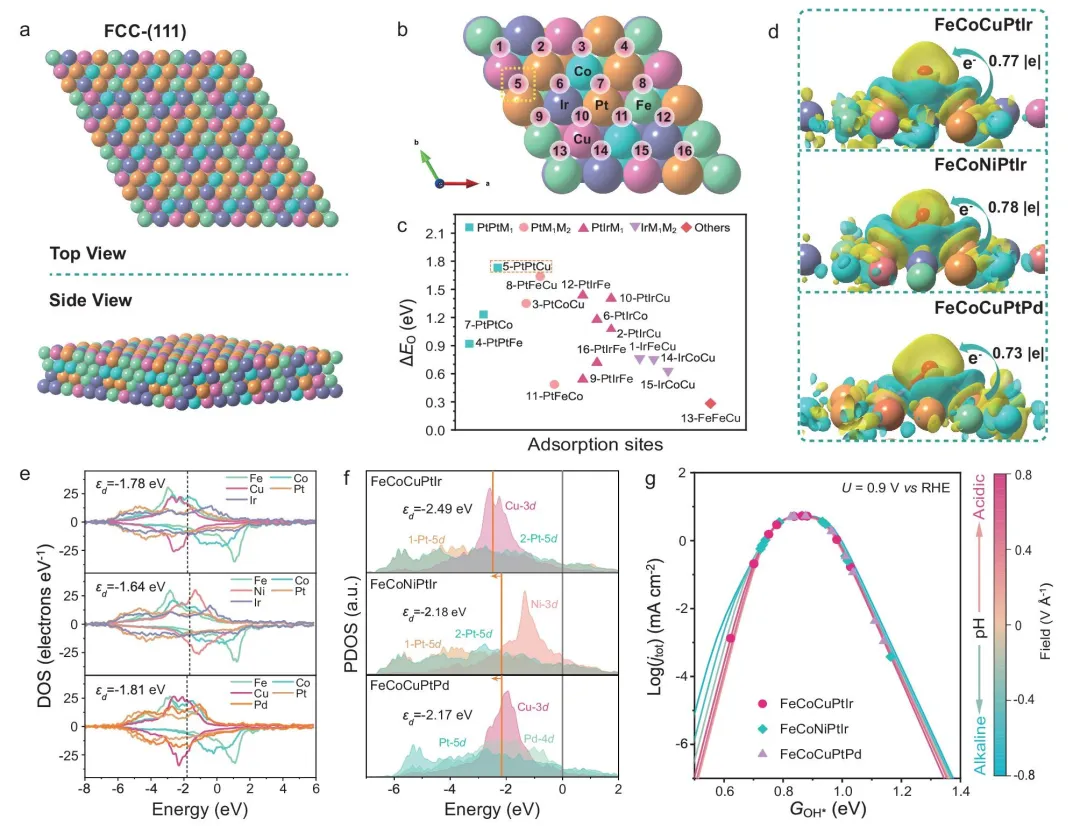
图 5|高性能 HEA 组合上 ORR 的机理分析。(a) FeCoCuPtIr–(111) 表面模型的俯视图和侧视图。(b) FeCoCuPtIr–(111) 表面上的 Hollow-FCC 吸附位点。(c) 相应的 ΔEO 数值。(d) O 吸附在三种 HEA 表面上的差分电荷密度分布。(e) 三种 HEA 组合的态密度图。(f) 三种 HEA 表面代表性活性位点的投影态密度图。(g) 在不同 pH 和电场条件下,0.9 V vs. RHE 时整体 ORR 的 pH 依赖微观动力学火山模型。
电子态密度分析进一步说明,Fe、Co、Ni、Cu 的引入能够调节 Pt 相关活性位点的电子结构,使 d 带中心整体下移,从而适度削弱 O* 与表面的相互作用,优化中间体吸附强度,促进 ORR 动力学。
在 pH 依赖 ORR 微观动力学火山模型中,FeCoCuPtIr 的 GOH* 分布中心最接近最优值,意味着其表面具有更高比例的“近最优”活性位点。这从统计意义上解释了 FeCoCuPtIr 在实验中展现出更优 ORR 活性的原因。
这项工作的意义:从 AI 辅助发现到闭环材料研发
这项研究的价值并不只是发现了几个高活性的 ORR 高熵合金催化剂,更重要的是建立了一种可推广的 AI 驱动材料发现范式。
在这个框架中,大语言模型不再只是用于问答或文献总结,而是被嵌入到真实科研流程中:它可以整合文献知识、辅助组合设计、生成实验方案、处理高通量数据,并进一步帮助研究者发现复杂材料体系中的隐藏规律。高通量实验平台则负责快速验证 AI 生成的候选组合,DFT 和微观动力学模型进一步提供机理解释。
这种“AI 知识推理—高通量实验验证—理论机制反馈”的协同模式,正是未来自动化材料实验室和数字材料生态的重要雏形。对于高熵合金这样的复杂多元素体系而言,它提供了一条从经验筛选走向数据驱动、知识增强和机制可解释设计的新路径。
当然,作者也指出,当前框架仍有提升空间。例如,目前尚未完全引入主动学习策略,也尚未实现候选材料的智能优先级排序;样品规模也受到合成通量、表征效率和筛选速度的限制。未来,如果进一步结合多模态 AI、贝叶斯优化、强化学习、自动化表征平台和标准化材料数据库,有望逐步形成真正闭环的“理性设计—自动实验—数据反馈—模型更新”材料创新系统。
总体而言,这项工作展示了 AI Agent、高通量实验和理论建模在复杂电催化材料发现中的深度融合,也为高熵合金催化剂的系统设计与机制理解提供了新的方法学基础。
论文信息:
X. Shan, F. Cai, Y. Tu, D. Zhang, Y. Pan, L. Chen, J. Liang, H. Gao, J. Xu, H. Li*, J. Li, E. Wang*. and M. Zhou*, "Unveiling the correlation between high-entropy alloy element systems and electrocatalytic activity", National Science Review, 2026, 13, nwag161.
链接:https://doi.org/10.1093/nsr/nwag161
