夜雨聆风
夜雨聆风





医疗器械设计开发合规指南:流程逻辑与文档要求 – 上期(医械人必备实操干货)

众多医械研发人员对设计开发各阶段文档的逻辑与顺序存在困惑,这直接关系到研发过程的真实性、可追溯性与合规性。相关文档既不应提前编制,也不应事后补做,必须遵循合规流程同步形成。
为帮助大家理清思路,医械加油站依据GB/T 42061-2022 / ISO 13485:2016及《医疗器械生产质量管理办法》,系统梳理设计开发核心阶段工作内容与合规要点,全文均为实操导向内容,建议收藏备用。
一、设计开发策划阶段:夯实合规基础,明确全流程框架
策划阶段是设计开发的顶层规划,核心是完成前期准备与整体部署,保障后续工作有序推进、风险可控。

1. 市场调研与可行性分析
本阶段需全面评估产品的市场价值与技术可行性,明确产品定位与实现路径,重点开展以下判断:
技术可行性:现有技术路线能否支持产品稳定实现与批量生产;
2. 立项管理
经评估具备可行性与市场竞争力后,应正式启动立项程序,明确:
3. 设计开发计划书

计划书应结合临床调研、用户需求与市场反馈编制,明确终端用户对产品性能、操作、安全性等方面的要求,核心内容包括:
4. 设计开发评审
二、设计开发输入阶段:明确合规要求,筑牢研发根基
输入阶段的核心是将用户需求与法规要求转化为清晰、可执行的设计依据,确保研发方向准确、合规。
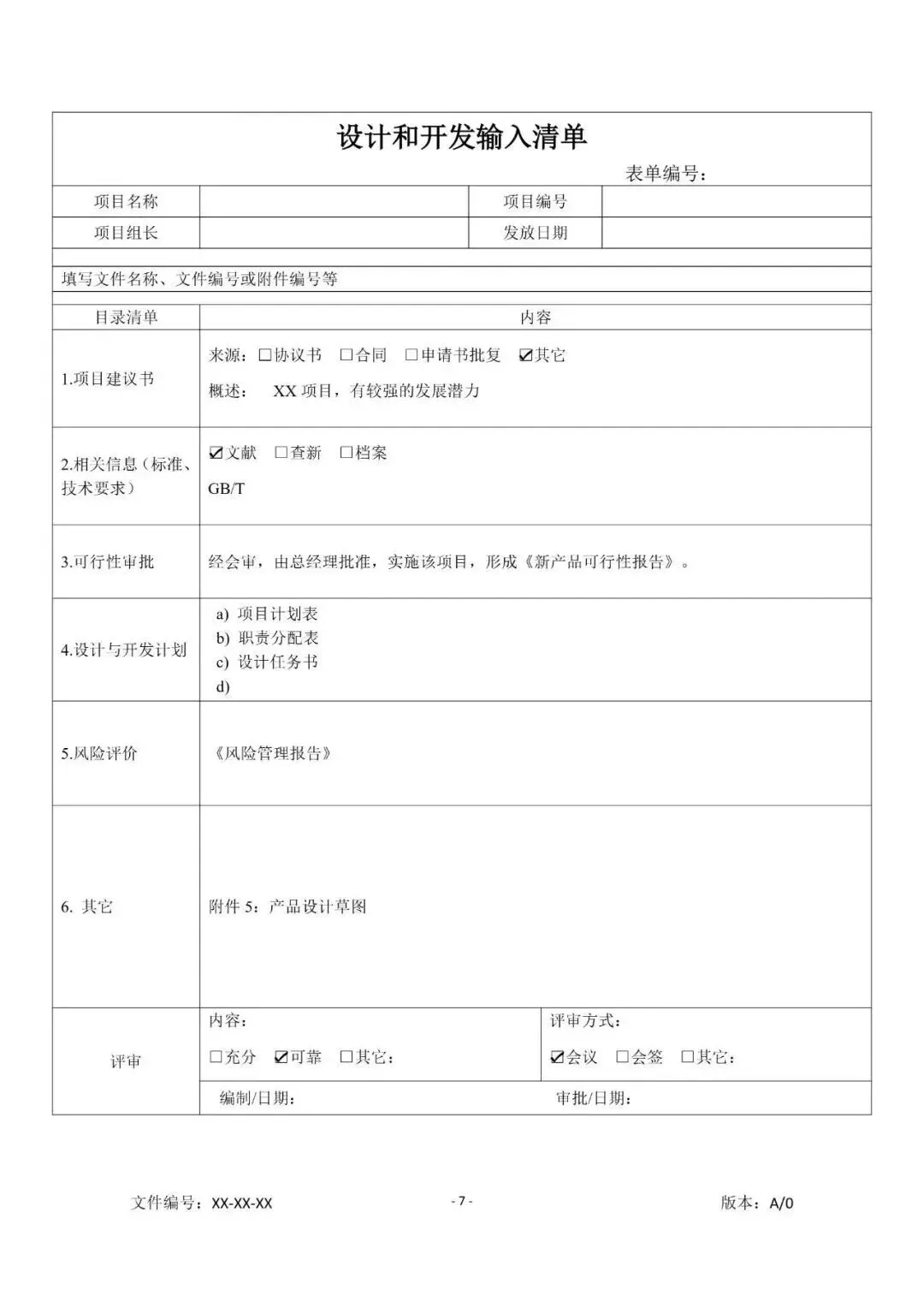
1. 用户需求规范
基于临床需求与预期用途,明确产品功能、性能、安全与人机工程要求,同步梳理适用法规与标准清单,确保需求既满足临床使用,又符合监管规定。
2. 风险管理输入
启动风险管理活动,全面识别与产品相关的潜在危害,包括生物相容性风险、电气安全风险、数据安全风险、使用错误风险、储运风险等,形成初始风险分析文件与风险管理计划,将风险控制贯穿研发全过程。
3. 设计开发评审
对用户需求与风险管理输入进行评审,确保输入完整、明确、无歧义,能够为后续设计开发提供稳定可靠的依据,避免因输入缺陷导致后期返工。
三、设计开发前段输出阶段:规范文档输出,保障全程可追溯
输出阶段需形成系列合规文件,作为生产、检验、注册、上市后的核心依据,所有输出应完整、可追溯、可验证。

1. 产品技术要求
作为前段输出的核心文件,应明确产品各项性能指标与检验方法。若无适用行业标准,可先行编制草稿版本,在研发过程中逐步完善并升版,同时保留完整版本记录。
2. 采购相关文件
输出关键原材料、组件与包装材料的采购技术要求及质量标准,明确技术指标、验证方法与接收准则,指导供应商筛选与物料验收。
3. 设计与检验文件
同步输出物料清单(BOM)、结构图纸、电气原理图、软件架构图等设计文件;编制原辅料、中间品、成品的质量标准与检验规程,为质量控制提供依据。
4. 工艺相关文件
输出工艺规程、工艺流程图等文件,明确关键工序与特殊过程,制定工艺研究方案,明确物料、设备、操作步骤、参数范围、判定准则与异常处理方式,保障工艺研究可重复、可追溯。
5. 软件相关文件(如适用)
含软件源代码、编译说明、版本记录、详细设计说明、用户界面设计等,确保软件研发过程合规可追溯。
6. 其他关键输出
包括产品标签、包装设计、说明书草稿等,均应符合对应法规要求。
7. 设计开发评审
输出阶段应开展多轮评审,验证输出的完整性、合规性与可实现性,确保能够支撑后续生产、检验与注册申报,对问题及时整改闭环。
四、设计开发核心原则:合规与可追溯为底线

设计开发全过程应严格遵循可追溯性原则,所有文件、研究记录、评审记录、变更记录均应完整留存,确保每一项设计、每一次调整均有据可查、逻辑一致、真实有效。
以上,我们重点梳理了非体外诊断试剂(IVD)类医疗器械设计开发中,策划、输入、输出三大核心环节的合规要点与实操细节,帮助大家搭建研发基础框架、理清各阶段文档逻辑及合规要求。设计开发合规落地离不开后续关键环节支撑,下一篇我们将继续深入,详细讲解验证、转换、确认、变更的具体内容。
后续医械加油站将持续输出医疗器械设计开发、质量体系、法规标准、注册申报等干货内容,助力行业同仁合规、高效、少走弯路,欢迎持续关注。