 夜雨聆风
夜雨聆风





JACS | 让 AI 看懂晶体: CrystalX 打通单晶结构解析“最后一公里”
一块只有针尖大小的晶体,能告诉科学家一个分子的真实模样。
在化学和材料实验室里,单晶 X 射线衍射长期被视为确定原子结构的“金标准”。衍射仪采集完数据后,研究人员面对的并不是一张清晰的分子照片,而是一团由电子密度峰组成的三维“地图”:哪个峰是碳,哪个峰是氮?氢原子应该补在哪里?结构是否符合化学常识和晶体学规则?
对于经验丰富的晶体学家来说,这是日常工作;但对于自动化合成、高通量结晶和自驱动实验室来说,这一步却常常成为卡住效率的“最后一公里”。前端实验越来越快,后端结构解析却仍需要专家反复判断、精修和验证,难以真正进入无人化、高通量的闭环。

近日,来自上海交通大学、上海人工智能实验室、上海创智学院的联合团队提出了深度学习系统 CrystalX,试图让 AI 学会“看懂晶体”。相关研究论文《CrystalX: High-Accuracy Crystal Structure Analysis Using Deep Learning》正式发表于国际化学领域顶级期刊《美国化学会志》(Journal of the American Chemical Society, JACS),第一作者为 MIFA 实验室二年级博士生郑凯鹏(创智 24 级学生),通讯作者为 MIFA 实验室黄维然副教授(创智全时导师)、上海人工智能实验室钟翰森研究员和李玉强研究员(创智全时导师)。目前,CrystalX 已全面开放并完成多平台部署。
-
项目代码仓库地址:https://github.com/kaipengm2/CrystalX
-
模型权重:https://huggingface.co/Kaipengm2/CrystalX
-
论文链接:https://pubs.acs.org/doi/10.1021/jacs.5c21832
-
“言普·晶析”:https://crystalx.intern-ai.org.cn
从“模糊地图”到全原子结构
CrystalX 要解决的问题,可以形象地理解为:给 AI 一张粗略的三维电子密度图,让它自动判断每个位置对应什么原子,并补全那些在 X 射线衍射中往往不够显眼的氢原子。
传统自动化晶体学工具更多依赖规则、经验阈值和人工设计的判断流程。CrystalX 则换了一种思路:不再手写规则,而是从大量真实实验数据中学习晶体结构背后的几何规律。
具体来说,CrystalX 将粗略电子密度峰看作三维点云,利用等变 Transformer 建模峰与峰之间的空间关系。第一阶段,CrystalX 接收由 SHELXT 等常规软件生成的粗电子密度峰图,再通过等变 Transformer 建模三维峰—峰相互作用,输出重原子结构框架。第二阶段,模型根据原子周围的分子内和分子间环境,预测每个重原子应连接多少个氢原子。这一步看似只是“补氢”,实际上需要理解完整的晶体环境。氢原子往往位于分子外缘,是否存在、应加几个,不仅取决于分子内部连接,也可能受到邻近分子和氢键环境的影响。因此,CrystalX 在预测氢原子时引入晶体的对称性和周期性,在每个非氢原子周围3.2 Å 范围内纳入对称等价的邻近原子,让模型同时感知分子内键连和分子间相互作用。研究显示,这种联合建模方式相比只使用分子内部信息,带来了超过 7% 的性能提升。
这样一来,原本需要专家在软件中来回检查的过程,被转化为一个可自动运行的几何深度学习流程。
这也是 CrystalX 与传统 rule-based 方法的重要区别:它不是简单地根据电子密度强弱“猜元素”,而是学习原子间距离、角度、二面角、氢键环境等更复杂的三维结构模式。对于 C/N/O、P/S/Cl 这类电子密度相近、传统流程容易混淆的元素组合,这种几何理解尤其关键。
面向“未来”真实实验数据的实战检验
为了检验 CrystalX 是否真的能用于日常晶体学工作,研究团队没有只在模拟数据上测试,而是构建了一个来自 Crystallography Open Database 的大规模真实实验数据集,包含 51,334 组 X 射线衍射数据,覆盖有机、金属有机和无机晶体,涉及 83 种元素和 86 个空间群。
更严格的是,团队采用了时间外推的验证方式:用 2018 年以前发表的结构训练模型,用 2018—2024 年发表的 8,834 个结构进行测试。这相当于让模型面对“未来的新题”,而不是在相似结构上重复刷题,更接近真实部署场景。
结果显示,CrystalX 在非氢原子识别上的原子级准确率达到 99.71%,在氢原子判断上的准确率达到 99.42%。如果用更苛刻的标准来看——一整个晶体结构中所有原子都必须判断正确,CrystalX 对非氢原子的结构级准确率达到 94.17%,对氢原子的结构级准确率达到 91.79%。此外,模型还能输出较为可靠的预测概率;利用这一不确定性信息,仅对最不确定的原子进行一次候选修正,就能将非氢原子和氢原子的结构级准确率进一步提升至 95.80% 和 94.35%。这组指标的意义在于,晶体结构解析不是“多数原子对了就行”。一个关键原子错了,可能就会影响后续精修、结构解释乃至化学结论。因此,结构级准确率更接近真实科研中的使用标准。
越是难题,越见稳定:
CrystalX 应对低质量数据和大型结构
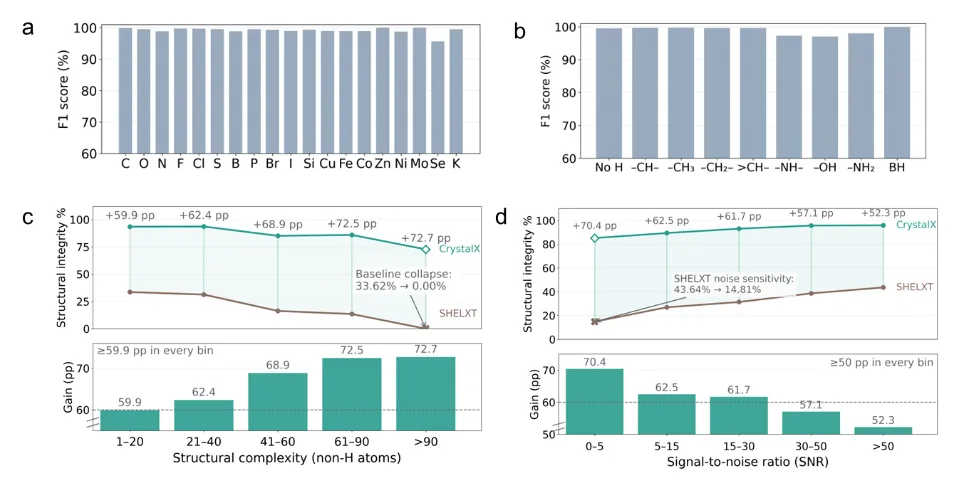
图:在大规模真实实验数据的时间外推测试中远超现有自动化工具,且在困难场景中的优势更为明显。
研究还将 CrystalX 与日常晶体学工作流中广泛使用的 Olex2 全自动流程进行了对比。基线流程包括:由 SHELXT 完成初步定相和非氢原子指认,通过 SHELXL 精修,然后使用 Olex2 的 hadd 命令自动加氢。整个过程不引入人工干预,因此可以代表当前常规晶体结构解析中的自动化水平。
在完整测试集上,SHELXT 对单个非氢原子的识别准确率达到 94.81%,看起来已经不低;但当标准提升为“整个结构中所有非氢原子都必须正确”时,结构级准确率降至 46.26%。相比之下,CrystalX 将这一指标提升至 94.17%,提高 47.91 个百分点,几乎实现了结构级准确率翻倍。
差距在困难场景中更加明显。面对低信噪比衍射数据,Olex2 自动流程仅正确解析 2/37 个结构,而 CrystalX 正确解析 24/37 个;面对大型复杂结构,Olex2 未能给出任何完全正确的结构,CrystalX 则成功解析 22/43 个。特别地,对于多达370 个非氢原子的庞大有机金属结构,CrystalX 仍能做到完全正确的秒级解析。
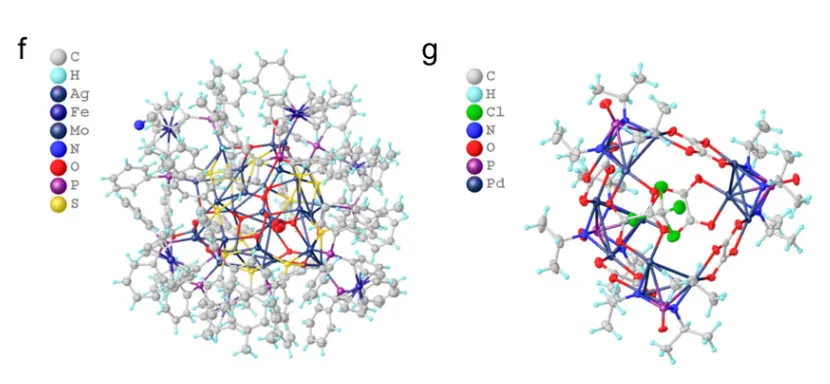
图:实现复杂结构的完全正确解析
这说明,CrystalX 的价值不只是“在简单题上更快”,而是在低质量数据、大型结构、元素组成复杂等传统自动化流程容易掉链子的场景中,仍然保持了更强的鲁棒性。
给已发表结构做“体检”:
CrystalX 发现顶刊当中隐藏的解析错误
更有意思的是,CrystalX 还被用来检查已经发表的晶体结构。
在测试集中,研究团队筛查了 1,559 个发表在 JCR Q1 期刊上的结构,自动过滤出 10 个可疑案例。经过人工复核,其中 9 个被确认为专家解释错误。这些错误包括相近电子密度原子的误判、氢原子放置错误、氢原子缺失。相比原始结果,CrystalX 给出的解析不仅在晶体学指标上表现更优,也在化学结构合理性方面更具说服力。
值得注意的是,部分错误并未触发 CheckCIF 的 A/B 类警报,而这通常已被视为相当严格的晶体学发表标准。换言之,这些结果不仅通过了严格的软件审查,也经受了论文发表过程中的同行评议,却仍然能够被 CrystalX 进一步识别并纠正。
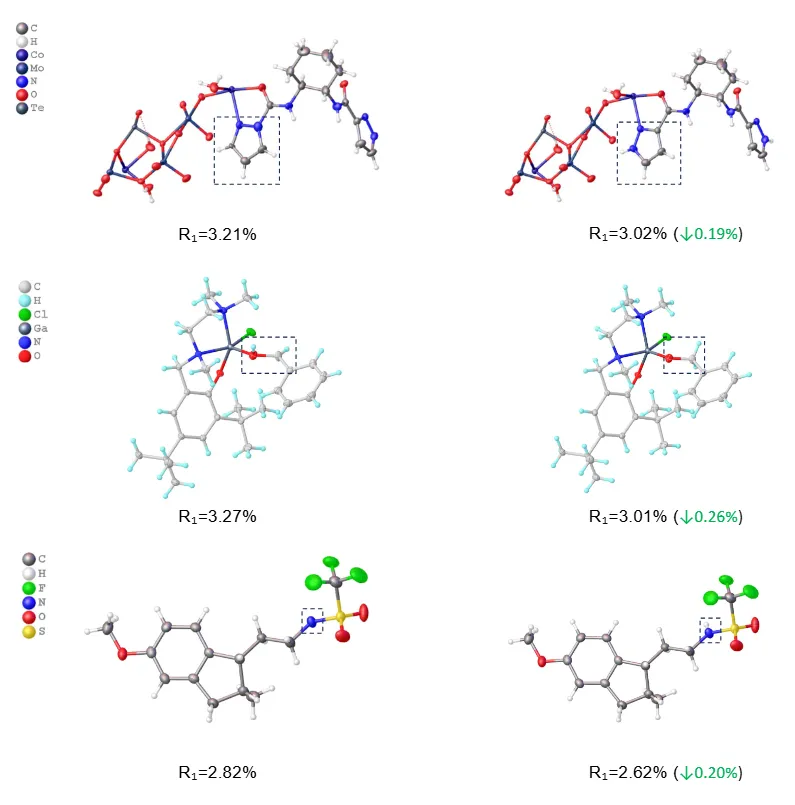
图:揭示顶刊论文中隐藏的解析错误
这让 CrystalX 的角色不再只是“自动结构解析工具”,也可以成为晶体学结果的智能复核助手。对于日益增长的结构数据库和高通量实验结果来说,这种能力具有现实意义:AI 不仅帮助科研人员生成答案,也能帮助他们发现答案中的错误。
从数据集评测到实验室闭环,CrystalX 迈向真实应用
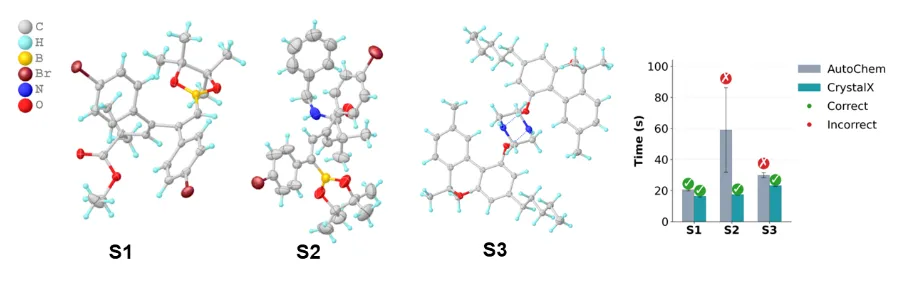
图:在真实实验中成功解析新发现化合物,超过AutoChem
为验证 CrystalX 在真实实验场景中的可用性,研究团队进一步将其接入日常晶体学工作流,并与具有代表性的全自动结构解析系统 AutoChem(最新版本 ac7)进行了前瞻性对比。
相比之下,CrystalX 的一个实际优势在于,它并不依赖这些仪器设置元数据,也不需要事先精确知道每种元素的组成信息。模型可以从常规软件自动生成的粗略电子密度峰出发,直接预测非氢原子类型和氢原子信息,再接入后续 SHELXL 精修与 CheckCIF 验证流程。这使得 CrystalX 更容易嵌入不同实验室已有的晶体学软件生态,也更适合未来高通量和无人化实验场景。
研究团队直接使用日常晶体学实验流程中的两个新发现化合物,并通过广泛文献检索加入一个近期发表且可运行AutoChem 的案例,构成三个真实应用样例。结果显示,CrystalX 在三个案例中均完成了正确的全自动结构解析。其中,两个新化合物由 CrystalX 生成的 CIF 文件没有 CheckCIF A/B 级警报,关键晶体学指标也处于合理范围内;随后,晶体学专家又通过传统精修流程独立确认了模型预测结果,二者保持一致。这表明,CrystalX 的解析结果已直接达到晶体学发表标准。这两个由 CrystalX 解析的新结构也已经提交至 Cambridge Crystallographic Data Centre。相比之下,AutoChem 在多种配置下最多只正确解析其中一个结构。这表明,CrystalX 的能力并不局限于数据库上的统计指标,而已经具备进入实际实验流程的潜力。
开源、可用,也面向下一步
目前,CrystalX 已全面开放并完成多平台部署。项目代码已在 GitHub 开源,仓库地址为:https://github.com/kaipengm2/CrystalX;模型权重已发布至 Hugging Face:https://huggingface.co/Kaipengm2/CrystalX。同时,CrystalX 已上线国家高等教育智慧教育平台(https://higher.smartedu.cn)、Migo 觅果化学助手(https://migo-chem.intern-ai.org.cn)以及“言普·晶析”(https://crystalx.intern-ai.org.cn),实现了最小端到端工作流的网页化部署。在该工作流中,用户只需上传初始 .ins 文件及对应的 .hkl 衍射数据,系统即可自动完成非氢原子识别、SHELXL 精修、氢原子预测与放置、进一步结构精修以及 CheckCIF 结构验证,并最终生成结果压缩包。压缩包内容包括 CIF 文件、CheckCIF 报告、SHELXL 相关文件以及模型预测概率等信息。此外,CrystalX 可自然接入现有晶体学软件生态,具备良好的可扩展性与可集成性,可作为实用晶体学工具服务于实际结构解析与精修流程。
现阶段,CrystalX 主要面向有序晶体结构的常规全原子解析,尚未覆盖晶体学无序这一更复杂的长尾场景。无序结构解析更像一道复杂“残局”:专家需要根据残余电子密度、占有率、约束条件和验证反馈,不断提出假设、精修、检查,再修正模型。这不是一次预测可以完成的问题,而是一个长时程序列决策过程。也正因如此,无序结构解析有望与当前快速发展的智能体 AI、强化学习等技术深度结合,使模型在与晶体学软件及相关工具的持续交互中,逐步学习并内化晶体学家在“解释—精修—验证”迭代过程中的决策机制。
总体而言,CrystalX 的意义不仅在于为单晶结构解析提供了一种新的深度学习工具,更在于它展示了一种有别于传统自动化晶体学流程的新范式:这种范式不再主要依赖人工设计的规则、经验阈值和启发式判断,而是通过大规模真实实验衍射数据学习粗略电子密度与原子结构之间复杂的几何关系,并据此直接推断全原子结构。相较于传统的 rule-based 方法,这一路径所代表的数据驱动几何深度学习框架展现出深刻的优越性。更重要的是,它有望从根本上改变当前晶体学分析对专家交互式操作的高度依赖,进而成为自驱动实验室和高通量化学发现流程中的关键自动化模块。
关于我们

MIFA 实验室全称 Machine Intelligence Foundations and Applications Laboratory,即机器智能基础与应用实验室,成员来自上海交大计算机学院和上海创智学院。实验室以长期的、有影响力的研究为驱动,致力于消除人工智能理论与实际应用之间的鸿沟,为 AI 的应用提供行之有效的理论指导。实验室团队小而精,科研氛围浓厚,负责人为黄维然副教授。目前,MIFA 实验室和多所知名高校、国家实验室、三甲医院以及业界大厂保持着紧密的学术合作与交流关系。