夜雨聆风
夜雨聆风
文章来源:momo读论文

论文信息
维度 | 信息 |
标题 | Deep contrastive learning enables genome-wide virtual screening |
作者 | Yinjun Jia, Bowen Gao, Jiaxin Tan, et al. |
机构 | 清华大学、北京智源人工智能研究院、北京大学等 |
论文地址 | https://www.biorxiv.org/content/10.1101/2024.09.02.610777v3 |
代码地址 | https://github.com/bowen-gao/DrugCLIP |
发表时间 | 2026年1月8日 |

一句话概要

背景与问题

核心贡献

方法详解
1. ProFSA 预训练(让塔台先看懂跑道)
ProFSA(蛋白质片段-周围区域对齐)的核心洞察在于:蛋白质内部的氨基酸相互作用与蛋白质-配体间的相互作用共享相同的物理化学法则,如氢键、π-π堆积等。研究团队从 PDB 蛋白质数据库中提取连续的短肽片段(1-8 个残基)作为“伪配体”,将其周围区域作为“伪口袋”,构建了 550 万个合成训练对。通过对比学习,让口袋编码器学会识别什么样的化学环境更容易结合分子。
2. 双阶段对比学习(给塔台装上一双慧眼)
DrugCLIP 包含两个编码器:分子编码器(初始化自 Uni-Mol)和口袋编码器(ProFSA 预训练)。训练目标是对比损失:
简单说,就是让真正的“口袋-配体”对在向量空间中靠近,让错误配对的远离。微调阶段使用 BioLip2 数据库中的 4 万多个实验复合物,并通过 RDKit 随机采样配体构象进行数据增强,让模型学会应对真实筛选时配体构象未知的情况。
3. GenPack 口袋优化(给不完美的跑道画线)
针对 AlphaFold 预测结构中侧链不准确的问题,GenPack 先去除侧链仅保留主链,用生成模型生成配体分子“定位”,再重新装回侧链并用分子力学优化。这个“去侧链-生成-装回”的过程,不是为了提高结构精度,而是为了更准确地定位口袋区域。

关键图表解读
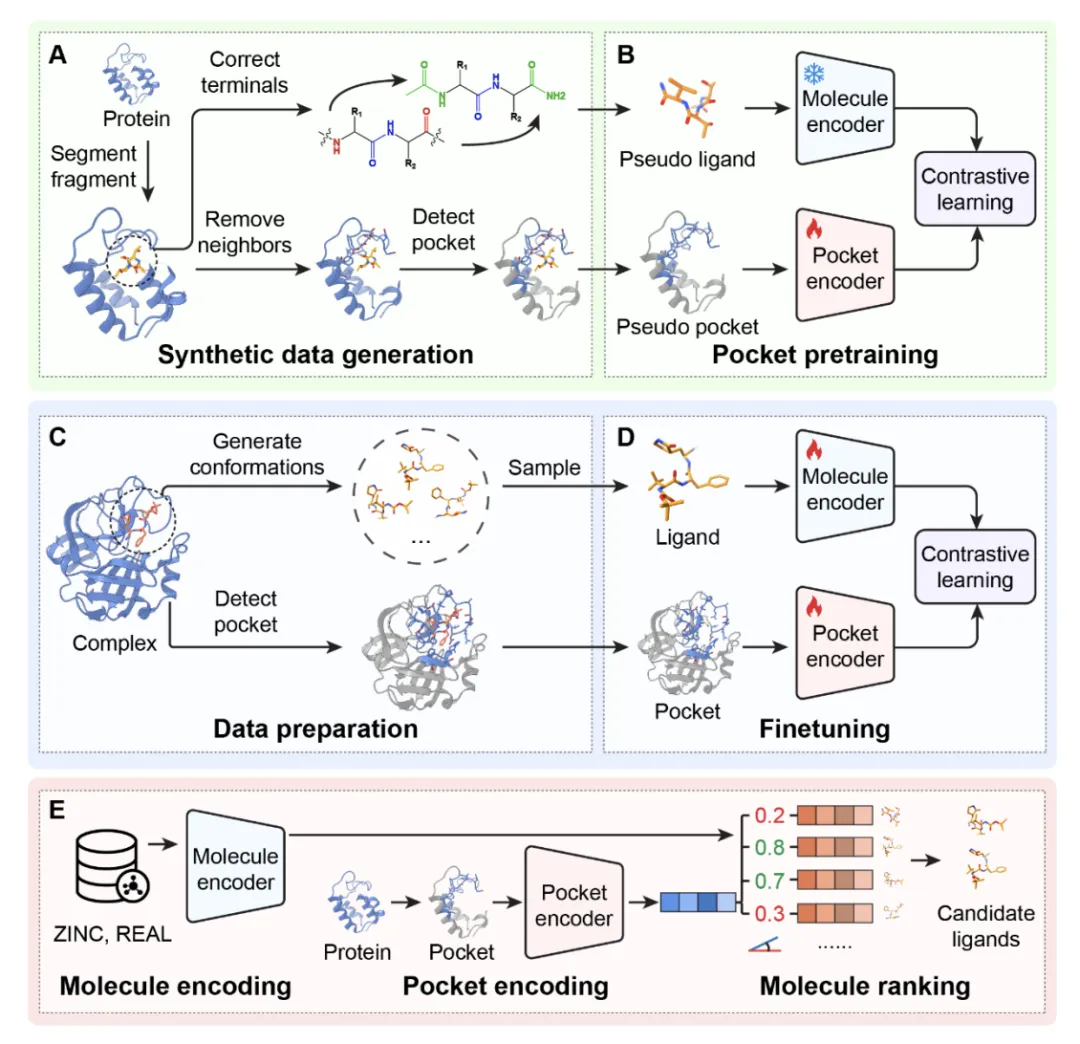
这张图完整展示了从预训练到应用的全流程。A图展示了ProFSA的核心:从蛋白质结构中切出片段(红色)作为伪配体,周围残基(蓝色)作为伪口袋。B图的对比蒸馏是关键创新:冻结优秀的分子编码器(Uni-Mol),让口袋编码器学习与之对齐。D图的微调阶段两者同时更新。E图的筛选流程最简洁:候选分子库一次编码、重复使用,对新靶点只需计算一次口袋向量,然后全部用余弦相似度排序,这是速度提升的核心。
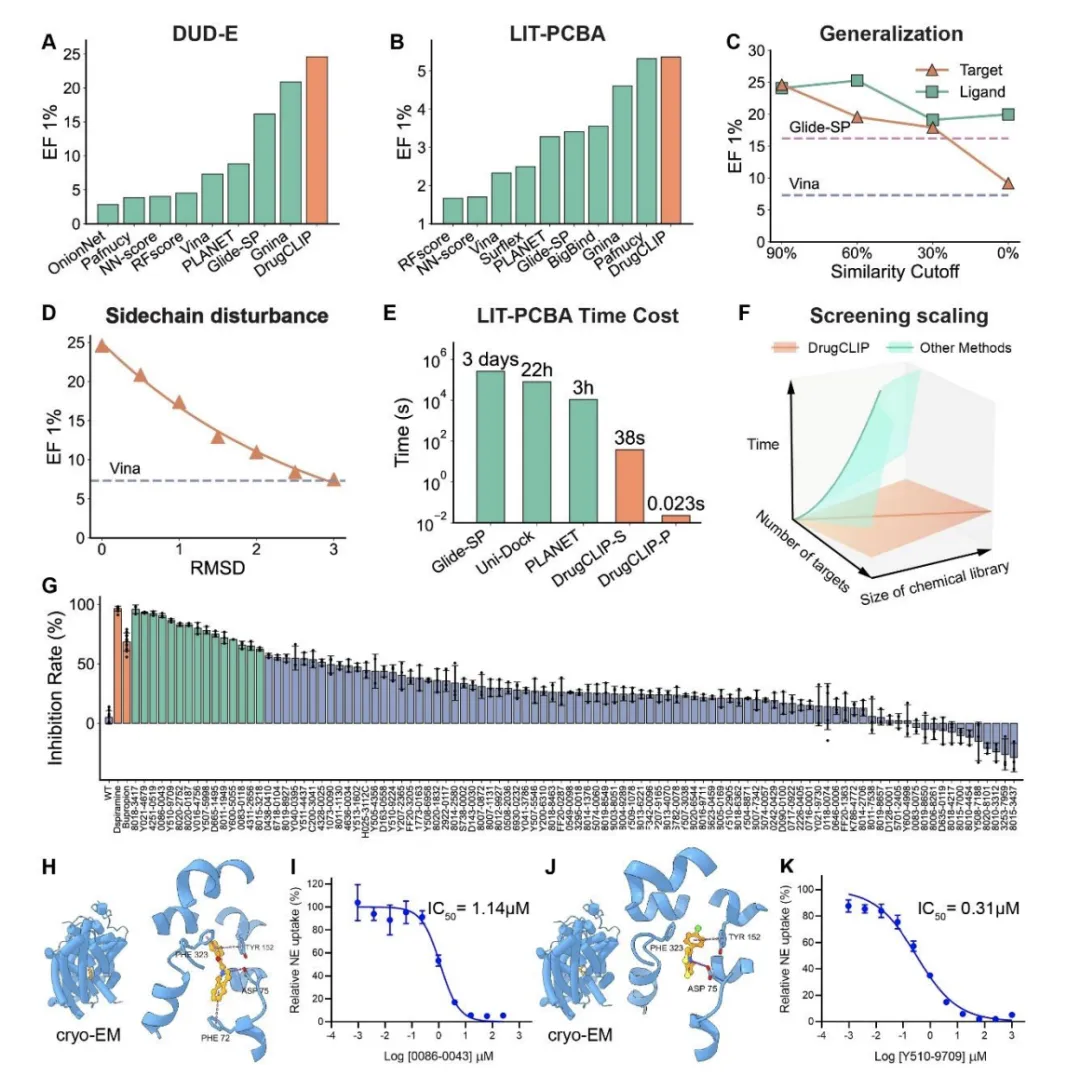
这张复合图信息量巨大。C图显示,即使移除所有与测试集共享相同Murcko骨架的分子(最严格的新颖性测试),DrugCLIP(EF1%≈20)仍优于Glide-SP(虚线≈16)。 D图更关键:当口袋侧链RMSD误差达到3Å时,DrugCLIP性能几乎不受影响,而AutoDock Vina(虚线)已大幅下降。这意味着模型对结构预测误差的容忍度极高。 E/F图的复杂度分析揭示本质:传统方法复杂度O(MN),DrugCLIP仅O(M+N),相当于从逐个面试变为集体简历筛选。
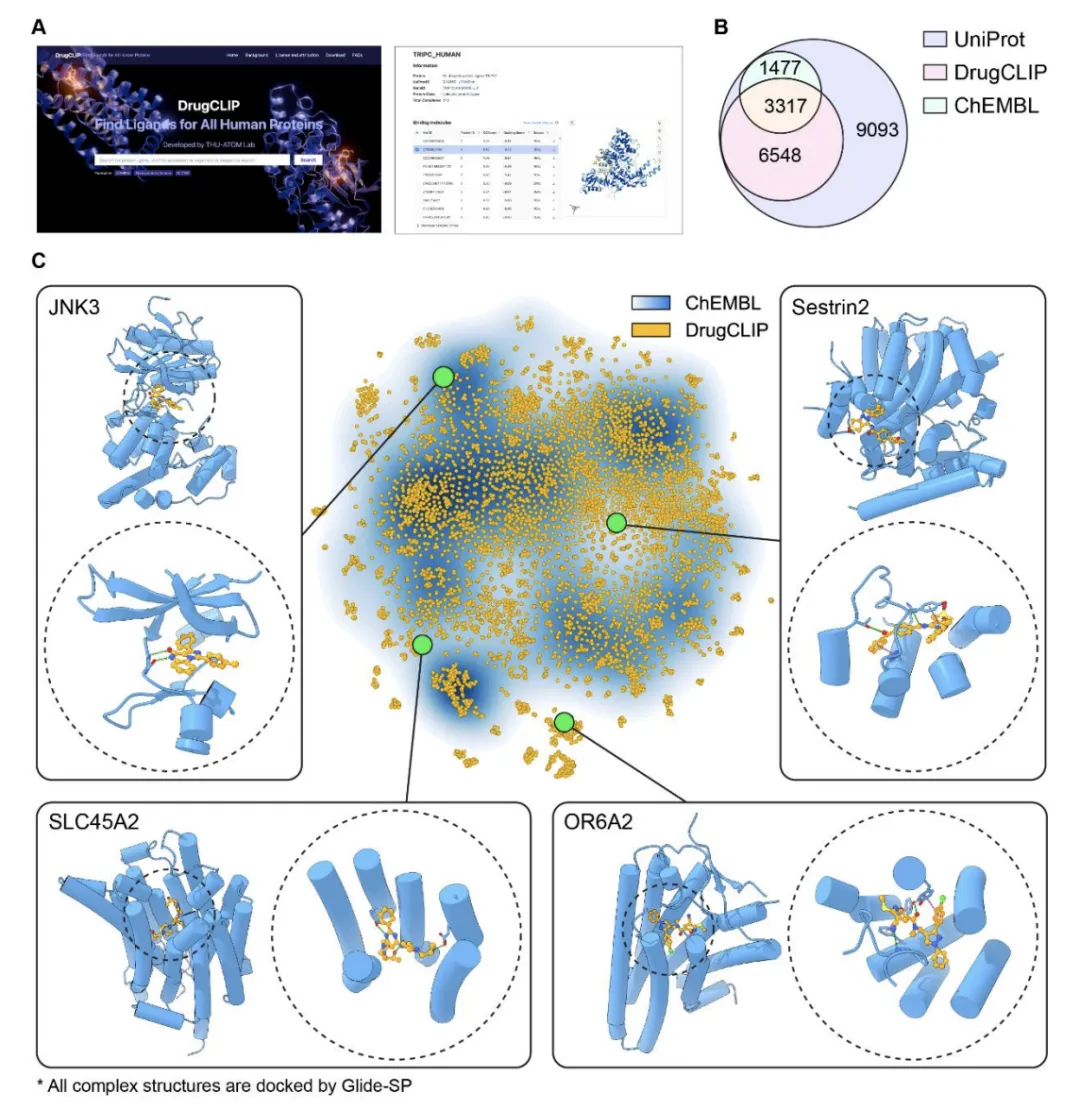
这是结果的最终呈现。B图Venn图显示,DrugCLIP覆盖9,908个人类靶点,是ChEMBL(4,810)的两倍多。C图的t-SNE可视化揭示了更重要信息:黄色(DrugCLIP覆盖)不仅包围蓝色(ChEMBL覆盖),还大量分布在蓝色稀疏区域——那些是迄今药理学理解有限的蛋白质家族,如嗅觉受体OR6A2(动脉粥样硬化新靶点)、Sestrin-2(癌症耐药相关)等。模型不仅找到了配体,还指明了这些“暗基因组”的潜在成药口袋。

实验与结果

优势与局限性
速度-精度不可三角被打破:同时实现SOTA精度、极端速度和强泛化性 对结构误差容忍度高:适合直接应用AlphaFold预测结构 真正的端到端基因组筛选:从靶点序列到候选分子一站式解决
目前仅针对单口袋筛选,无法处理变构调节或多口袋协同 GenPack生成的分子不一定可合成,仅用于定位口袋 湿实验验证主要集中在少数靶点,全基因组范围的假阳性率仍需大规模实验检验

结论与启发
对于那些连AlphaFold都预测不准的无序蛋白区域,我们又该如何设计筛选策略? 当化学空间从5亿扩展到50亿(如Enamine REAL Space),DrugCLIP的检索效率和精度还能保持吗?