 夜雨聆风
夜雨聆风01 引言:分子动力学进入“从头算”精度时代
分子动力学(MD)模拟是现代生物物理、化学和材料科学的基石。在过去的三十年里,GROMACS 凭借其极致的计算性能和出色的平台可移植性,已成为分子模拟领域的“行业标准”。然而,传统的经典 MD 模拟高度依赖于经验力场(如 AMBER、CHARMM),虽然计算效率极高,但在处理电荷转移、化学键断裂以及多体极化等量子效应时往往力不从心。
近年来,机器学习原子间相互作用势(MLIPs),特别是以 DeePMD-kit 为代表的深度势能(Deep Potentials)技术,为这一难题提供了突破性的解决方案。它们通过深度神经网络拟合量子力学(QM)数据,实现了接近从头算(Ab-initio)的精度,同时保持了接近经典力场的计算效率。
然而,如何将复杂的神经网络推理高效地嵌入到 GROMACS 这种经过极致优化的传统引擎中,并实现在大规模多 GPU 集群上的并行扩展,依然是高性能计算(HPC)领域的重大挑战。来自瑞典皇家理工学院(KTH)的研究团队近日在 arXiv 上发表了最新成果,详细介绍了他们如何通过扩展 GROMACS 接口,实现了与 DeePMD-kit 的深度集成,并成功在 NVIDIA 和 AMD 顶级 GPU 上完成了万级原子规模的高效模拟。
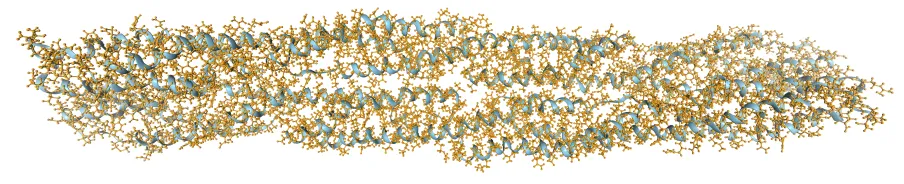
Figure 1:展示了包含 15,668 个原子的 1HCI 蛋白质系统。这是本研究用于性能测试的核心系统,证明了 GROMACS-DeePMD 耦合方案在处理大规模生物大分子时的稳定性。
02 深度势能模型:从 DP-SE 到 DPA-1 的进化
在深入集成方案之前,我们需要理解深度势能模型的核心架构。DeePMD-kit 采用的是一种“描述符(Descriptor)+ 拟合网络(Fitting-net)”的范式。
2.1 描述符与拟合网络
描述符负责将原子及其局部环境(截断半径内)的信息编码为保持平移、旋转和排列对称性的矩阵。拟合网络则是一个多层感知器(MLP),将这些特征映射为单个原子的能量。系统的总能量即为所有原子能量之和。
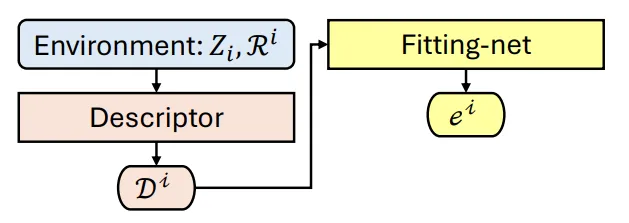
Figure 2:描绘了从原子类型、位置信息到描述符生成,再到最终能量预测的完整计算流。
2.2 为什么选择 DPA-1?
研究团队重点讨论了四类深度势能模型:
1. DP-SE:经典的平滑版深度势能,应用最为广泛。 2. DPA-1:引入了门控自注意力(Gated Self-attention)机制,能更精准地捕捉原子间的相关性。 3. DPA-2 / DPA-3:属于“大原子模型(LAMs)”,引入了消息传递机制,允许信息跨越多个“跳数”传播。
在本项工作中,团队选择了 DPA-1。这是因为 DPA-1 具有“局部性(Locality)”特征——原子的受力仅取决于其局部邻居,不涉及跨中心的耦合。这种特性使其在分布式内存环境下的区域分解算法中更具优势,避免了消息传递模型带来的超大通信开销。
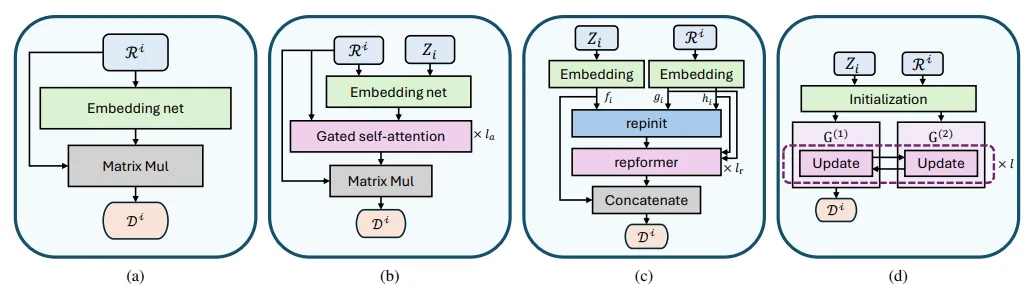
Figure 3:详细对比了不同代际模型的演进逻辑,展示了 DPA-1 如何通过注意力机制提升精度。
03 技术突破:解耦的区域分解策略
将 AI 模型集成到 GROMACS 最大的难点在于如何处理区域分解(Domain Decomposition, DD)。
在经典的 DD 模拟中,空间被划分为多个子域,每个 MPI 进程负责一个子域。为了计算跨域的相互作用,通常需要建立“影子原子(Ghost Atoms)”层。然而,神经网络势能对邻居信息的要求远比经典力场严苛,传统的通信模式往往会导致严重的性能损失。
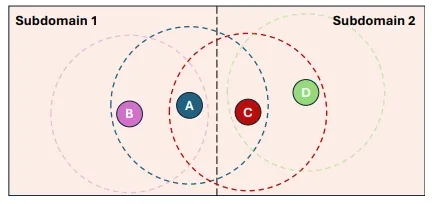
Figure 4:区域分解下的邻居列表依赖示意图
3.1 扩展 NNPot 接口
研究团队利用了 GROMACS 2025 预览版中引入的 NNPot 接口。他们开发了一个专用的 DeePMD 后端,该后端能够直接调用 DeePMD-kit 的 C++ API,实现高效的数据交换。
3.2 虚拟区域分解层
这是本项研究的核心创新:团队引入了一个与 GROMACS 主模拟循环完全解耦的临时虚拟区域分解层。
• 独立性:NNPot 内部的原子分布不需要与 GROMACS 主循环的 DD 一致。这使得系统在执行 AI 推理时,可以根据 AI 负载进行更均匀的分配。 • 高效通信:在每个模拟步长中,系统通过 MPI_Bcast将坐标广播到所有 Rank,推理完成后利用MPI_Reduce或MPI_Allreduce聚合力数据。这种策略极大简化了影子原子的管理逻辑,降低了代码实现的复杂度。
04 实验验证:精度与物理一致性
为了确保集成方案不仅快而且准,团队进行了严格的验证。
4.1 模型训练
团队针对溶剂化蛋白质片段训练了一个包含 160 万参数的 DPA-1 模型。训练结果显示,力(Force)的均方根误差(RMSE)达到了约 0.2 eV/Å,这与目前最先进的 DPA-2 模型的精度持平。
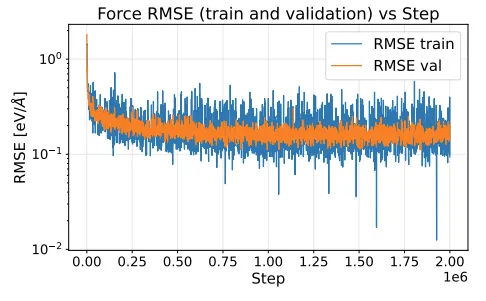
Figure 7:模型训练过程中力 RMSE 的演变曲线
4.2 稳定性测试
通过对 1YRF 蛋白质进行长达数纳秒的模拟,团队对比了 DPA-1 模型与传统力场下的回转半径(Radii of Gyration)。结果表明,AI 模拟下的蛋白质结构极其稳定,没有出现任何物理上的异常膨胀或崩溃,证明了虚拟区域分解算法的正确性。
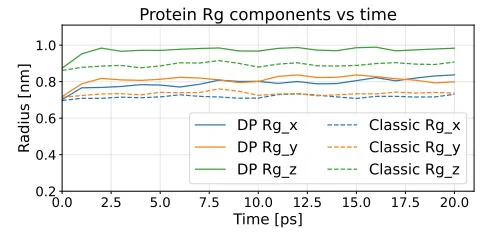
Figure 8:DPA-1 与经典力场模拟下蛋白质回转半径随时间的变化对比
05 性能大考:NVIDIA A100 vs AMD MI250x
性能测试在两台世界级的超级计算机上展开:配备 NVIDIA A100 的 Leonardo 集群和配备 AMD MI250x 的 LUMI-G 集群。
5.1 强缩放性能(Strong Scaling)
在 15,668 原子的 1HCI 系统测试中,集成方案展现了出色的扩展性:
• 在 NVIDIA A100 上,从 8 个 GPU 扩展到 32 个 GPU 时,效率保持在 40% 以上。 • 值得注意的是,AMD MI250x 在高 Rank 数(24-32 卡)下表现略优,这得益于其单节点内更多的计算单元(GCD)降低了跨节点 MPI 通信的压力。
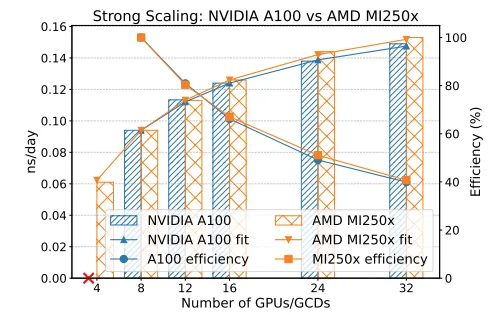
Figure 10:NVIDIA A100 与 AMD MI250x 的强缩放性能对比图
5.2 性能瓶颈分析
研究团队使用 ROCm System Profiler 进行了深度剖析,揭示了 AI MD 模拟的时间分配:
• 推理占统治地位:超过 99% 的墙钟时间消耗在 NNPot 模块中,即执行 DeePMD 的推理任务。相比之下,传统的 GROMACS 经典操作(如积分、坐标更新)耗时不到 1%。 • 同步开销:由于采用了 MPI 集体通信,力的聚合阶段(MPI Collectives)成为了全局同步点。如果不同 Rank 间的计算负载不均衡,最慢的 Rank 会拖慢整体速度。
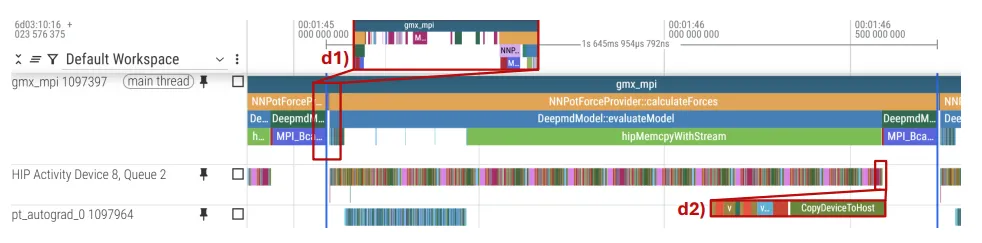
Figure 12:使用 ROCm Profiler 得到的 16 Rank 模拟时间线追踪图
06 总结与未来展望
本项工作成功地在 GROMACS 中为 AI 势能“腾出了空间”。这项研究的意义不仅在于提供了一个高效的工具,更在于其验证了一套可行的集成模式:
1. 跨平台支持:同时适配 CUDA 和 ROCm 环境,充分释放了当代超算的硬件潜力。 2. 生产级性能:通过 DPA-1 模型,研究者现在可以在保留量子级精度的前提下,对万级甚至十万级原子的蛋白质系统进行纳秒级的长时间模拟。 3. 架构参考:虚拟解耦的区域分解策略为未来集成更复杂的消息传递模型(如 DPA-2/3)提供了宝贵的经验。
随着 DeePMD-kit 与 GROMACS 融合的不断深入,我们有理由相信,AI for Science 将在生物制药、材料发现等领域发挥越来越关键的作用。
参考文献:Pennati, L., Hu, A., Peng, I., Müllender, L., & Markidis, S. (2026). Making Room for AI: Multi-GPU Molecular Dynamics with Deep Potentials in GROMACS. arXiv:2604.07276v1 .
