 夜雨聆风
夜雨聆风前面我们给大家介绍了R中的智能体 aisdk,见:
aisdk:在R console聊天,让AI干活!(活人版本教程)
aisdk还是太超前了:继Y叔,生信技能树被蒸馏后,还可以自主接入任何想要的skills!
然后在使用的过程中,我提了一个小的建议,见上面的稿子,你可以找到,我当时觉得我那个想法有点痴心妄想,但是!YH的脑子不知道怎么长的,转身就在aisdk里面加了一个新的功能 ask_ai(),使用过后,直接打开新世界,给你带来全新的体验!!!
来看看!
情景复现1:
我这里在本地电脑上面测试monocle2的代码,会遇到一些以前遇到的报错,原因也基本都知道怎么修改,但是为了测试 aisdk 的新功能 ask_ai(),我决定让这个R里面的智能体来给我修改!
测试数据:
在这里可下载:44-单细胞多组火山图 链接: https://pan.baidu.com/s/121YDbgxYuQHW-UZvlLdfsQ?pwd=cpwh 提取码: cpwh
首先运行R代码:
rm(list=ls())library(aisdk)library(monocle) # 单细胞轨迹分析核心包library(Seurat)library(dplyr)library(ggplot2)library(igraph)set.seed(42)getwd()dir <- "./5-monocle2/"print(getwd())# 1.读取已注释的Seurat对象----subset_data <- qs::qread("./4-subcluster_T-NKcells/sce.all_int.qs")subset_datahead(subset_data@meta.data)meta <- readRDS("4-subcluster_T-NKcells/phe.rds")subset_data <- AddMetaData(subset_data, metadata = meta)table(subset_data$celltype_sub)DimPlot(subset_data, label = T,group.by = "celltype_sub")Idents(subset_data) <- "celltype_sub"table(subset_data$celltype_sub)# 选择和抽样细胞子集# 选择特定细胞类型进行分析(纤维细胞、内皮细胞、上皮细胞)subset_data <- subset(subset_data, ident = c('CD8+ cytotoxic T'))table(Idents(subset_data))head(subset_data@meta.data)#aisdk::ask_ai(skill = "小树")# 2.Seurat元数据整合和Monocle对象创建----# 从Seurat对象提取count矩阵expression_matrix <- GetAssayData(object = subset_data, assay = "RNA", slot = "counts")这里出现了一个报错:
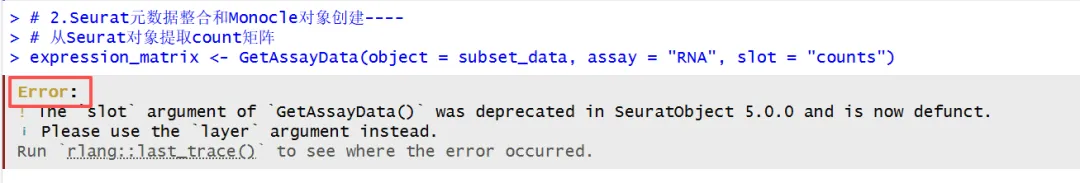
这个时候使用 aisdk::ask_ai() 进入报错诊断,接住上面的报错:
aisdk::ask_ai()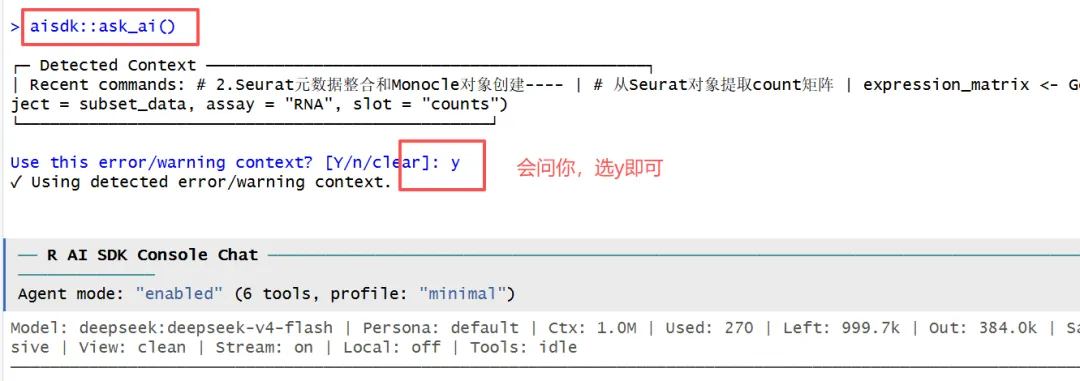
他这里诊断可能有点久,我就按了esc,重新给了他一句提示:
帮我修改这句代码变成可以提取count就行
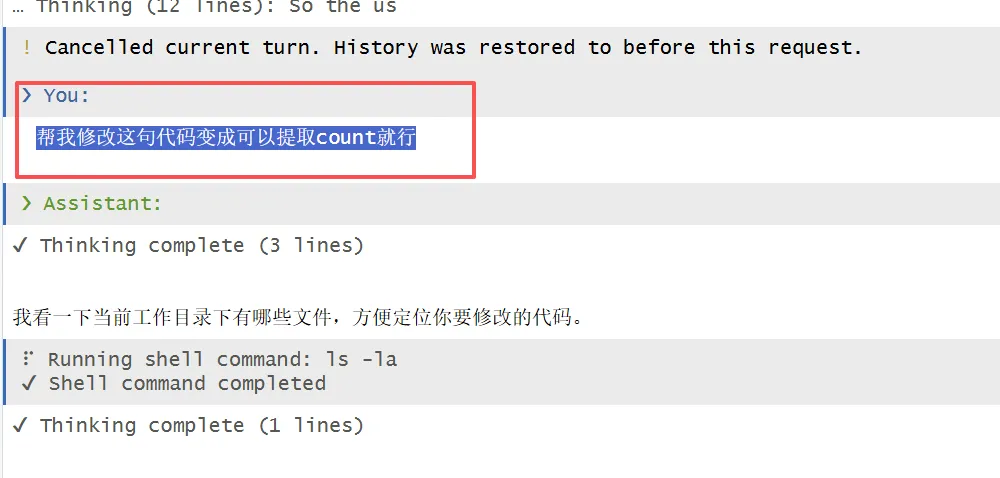
然后它直接修改了我的R脚本中的代码:

我重新跑了上面的,非常顺利丝滑!!!
情景复现2:
下面这个报错的解决方案是我始料未及的,它真的太棒啦!以前我都是自己手动修改monocle2的R包,但是这次 ask_ai() 给的方法很惊喜!!!
来看看!
上面monocle2的脚本接着运行:
# 2.Seurat元数据整合和Monocle对象创建----# 从Seurat对象提取count矩阵expression_matrix <- GetAssayData(object = subset_data, assay = "RNA", layer = "counts")# 准备细胞元数据cell_metadata <- new('AnnotatedDataFrame', data = subset_data@meta.data)# 准备基因注释信息gene_annotation <- new('AnnotatedDataFrame', data = data.frame(gene_short_name = row.names(subset_data), row.names = row.names(subset_data)))# 3.创建 Monocle 核心对象 (CellDataSet)----# expressionFamily = negbinomial.size:指定使用负二项分布处理UMI数据monocle_cds <- monocle::newCellDataSet(expression_matrix, phenoData = cell_metadata, featureData = gene_annotation, lowerDetectionLimit = 0.5, expressionFamily = VGAM::negbinomial.size())monocle_cds# 4.Monocle2 预处理与轨迹推断----# 4.1 数据预处理和归一化cds <- monocle_cds # 重命名对象便于后续操作cds <- estimateSizeFactors(cds) # 标准化测序深度差异cds <- estimateDispersions(cds) # 估计基因表达的离散度运行到这里,又是一个非常经典的报错!

跟上面一样,输入下面的命令接住上面的报错:
aisdk::ask_ai()他就开始检测我的报错内容了,然后问我是不是要用这个error,选择y即可:
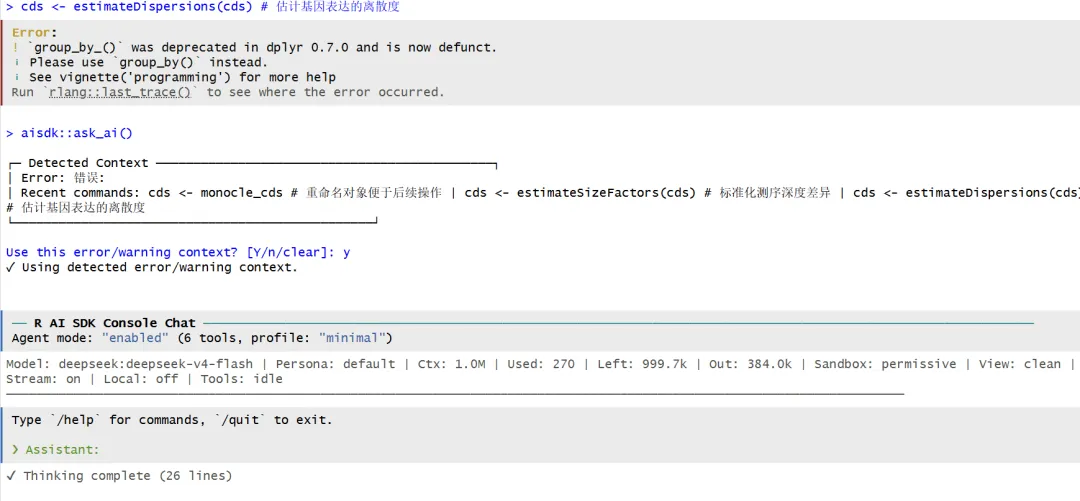
到这里它已经思考完了,但是我好像看不到细节,所以我让它给我看细节:
你可以展示你的思考过程吗,现在只显示多少行,看不到细节

上面给了很详细的解决方案,但是我们不必自己动手,我让它给我修补:
你帮我插入你的修复方案,然后插入的代码记得给我个标记知道是你插入的
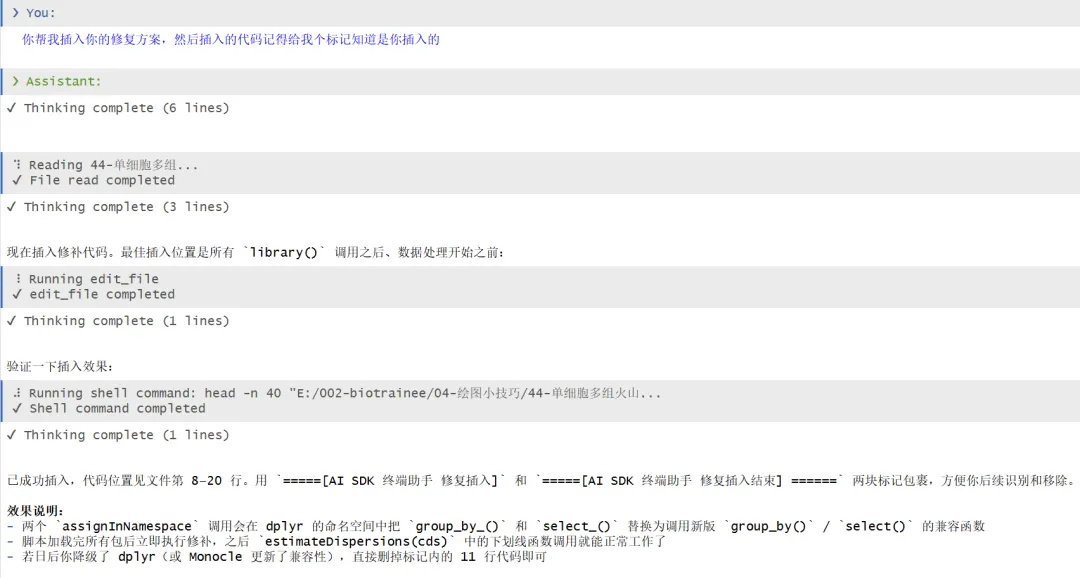
插入的一段代码:
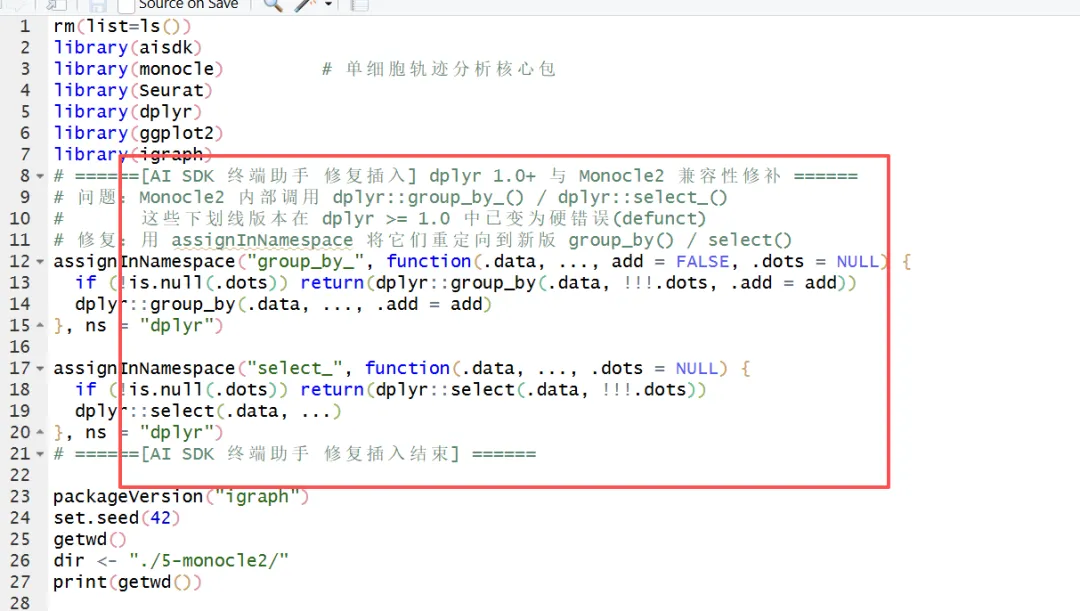
退出然后看看效果咋样:
/quit运行成功了!
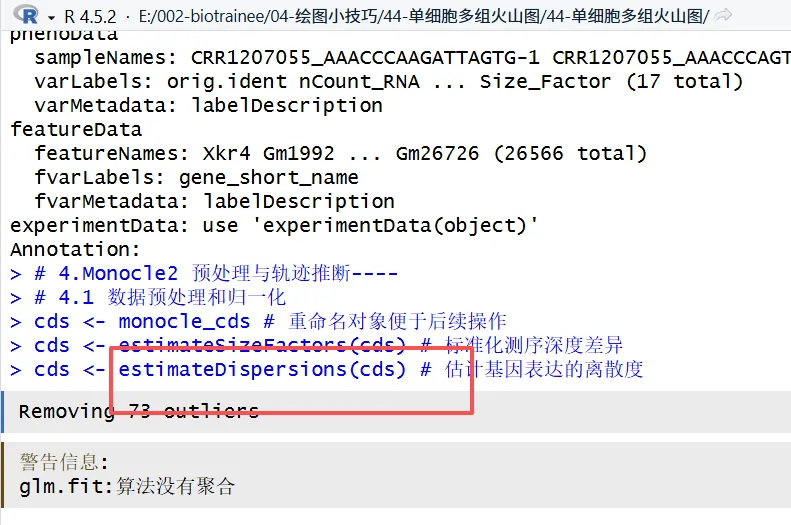
是不是超级棒!!!
R中的报错,解决方案直接给出新的世界打开方式!
AI交流,欢迎来群里交流玩,请备注AI交流:
友情转发:
AI加持,全新改版!生信入门&数据挖掘线上直播课5月班,系统的生信入门课 生信故事会,来看看他们的生信入门故事 生信马拉松答疑专辑,获取你的生信专属答疑 GEO数据实战训练直播(学员免收门票),课后有大量案例实战训练
