 夜雨聆风
夜雨聆风今天给大家分享一篇发表在《Chem》上的重磅研究成果。对于从事生物催化、酶挖掘以及结构生物学的小伙伴来说,这篇文献提供了一个极具启发性的全新范式——如何利用蛋白质语言模型(PLMs)和基于Motif的深度学习,在序列同源性极低的盲区中,精准挖掘出具有特定功能的全新酶。
1. 文献标题
A motif-based deep learning tool for the identification of unusual NADH-dependent imine reductases(一种基于基序的深度学习工具,用于鉴定不寻常的NADH依赖型亚胺还原酶)
2. 作者团队
作者:Xin-Yuan Shen, Yu-Xuan Wu, Zhi-Feng Ma 等
通讯作者:Gui-Sheng Fan, Gao-Wei Zheng
通讯单位:华东理工大学,生物反应器工程国家重点实验室
团队研究方向:生物催化、酶工程、结构生物学及生物制造。
3. 发表年份
2026年
4. 研究背景
亚胺还原酶(IREDs)是手性胺合成的核心生物催化剂,催化亚胺还原(IR)和还原胺化(RA)反应。然而,自然界中绝大多数已知的 IREDs 都严格依赖 NADPH 作为辅因子。NADH 相比 NADPH 具有价格更低廉、更稳定且更容易与现有代谢网络整合的优势。但目前天然的 NADH 依赖型 IREDs 尚未被开发。由于缺乏已知的靶序列,传统的基于序列比对(如 BLAST)的方法很难在基因数据库中挖掘出这类酶。
5. 研究目的
开发一种超越传统序列比对限制的新型生物信息学工具,以此在海量数据库中挖掘并鉴定出自然界中全新且天然偏好 NADH的亚胺还原酶家族,进一步扩充手性胺生物合成的工具箱。
6. 研究问题
在完全没有目标序列作为模板、且潜在目标蛋白与已知蛋白序列同源性极低的情况下,如何凭借“辅因子结合特征”,大海捞针般地找出隐藏在百万序列库中的新酶?
7. 研究的逻辑思路
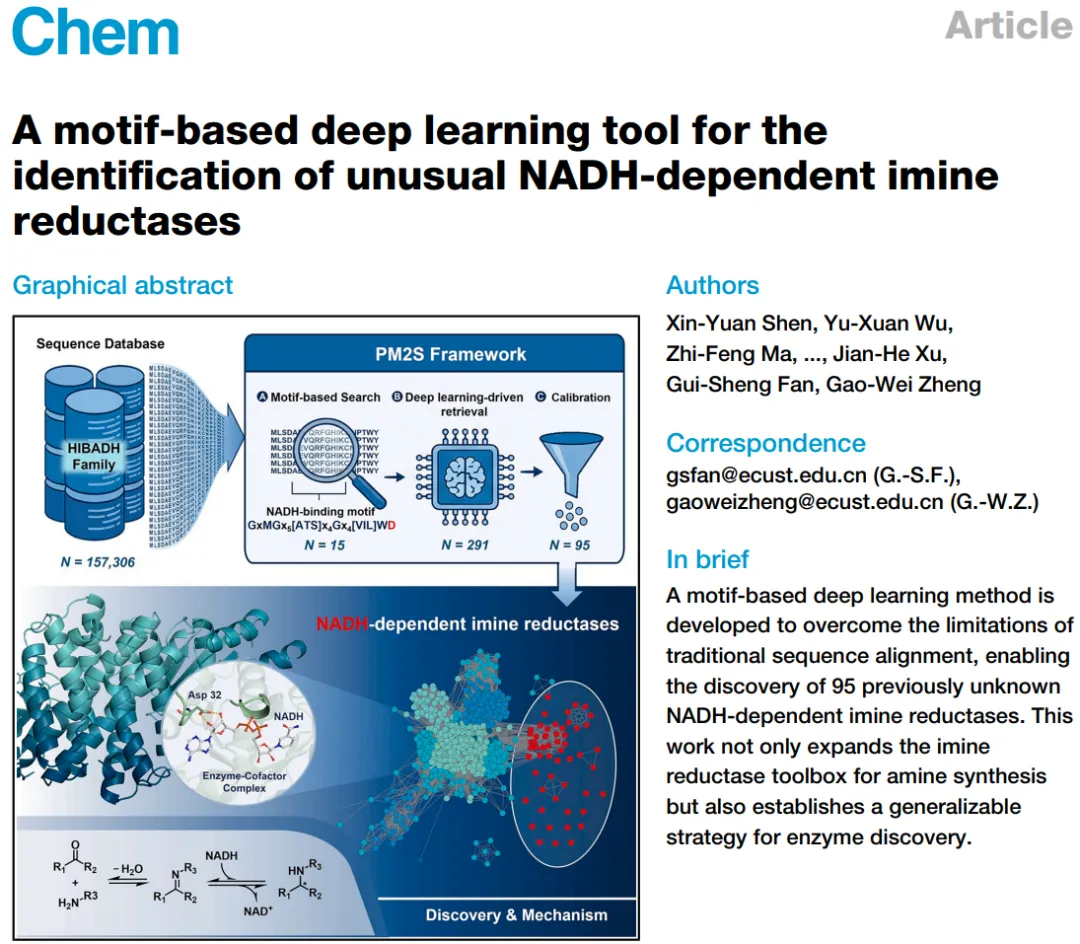
作者摒弃了“看全序列相似度”的老路,采用了一套名为PM2S (Protein Motif to Search)的“漏斗式”挖掘逻辑:
交叉借用,理性设计 Motif:从结构同源的$\beta$-羟基酸脱氢酶($\beta$-HADs)中汲取灵感,将 NADPH 结合基序中的关键精氨酸(Arg)替换为决定 NADH 偏好性的天冬氨酸(Asp),人为构建出一段“NADH 偏好结合基序”(NADH-binding motif)。
规则初筛获取“种子”:用该 Motif 在扩充的数据库中进行正则表达式匹配,抓取少量符合条件的“种子蛋白”。
大语言模型降维扩展:将种子蛋白和数据库序列转化为高维向量(Embeddings),在语义空间中进行深度的迭代检索,大幅扩充候选池。
规则反向校准:再次利用正则表达式,剔除掉含有典型 NADPH 结合基序的假阳性序列。
湿实验“盖棺定论”:通过多重底物活性筛选、产物转化及 AlphaFold/X射线晶体学解析酶-辅因子复合物的机制。
图 1. 发现假定的天然 NADH 依赖型 IREDs。 (A) PM2S 挖掘平台的核心工作流程;(B) IRED NADH 结合基序的理性构建原理;(C) 15 个种子蛋白的鉴定及以 NADH/NADPH 为辅因子时的活性比较。
8. 理论基础 / 分析框架
酶结构进化理论:Rossmann 折叠域对辅因子的选择往往由关键位点的单个氨基酸残基(如 Asp 或 Arg)决定,且该催化核心基序高度保守。
蛋白质语言模型(PLMs):蛋白质序列可以被理解为一种“语言”,深度学习模型(如 ESM1b)能将其编码为蕴含结构和功能特征的高维致密向量(Dense vectors),这使得即使序列一致性很低,也能通过计算向量相似度找到功能相似的蛋白。
9. 研究方法
计算生物学方法:整合了正则表达式(Rule-based)、动态规划、随机森林算法、预训练蛋白质大模型(ESM1b)以及 Milvus 向量数据库检索技术(PM2S 平台)。
分子生物学与酶学:基因合成、大肠杆菌异源表达、Ni-NTA 亲和纯化、稳态动力学分析、气相/液相色谱(GC-FID/HPLC)底物谱测试及半制备级生物转化。
结构生物学:AlphaFold3 复合体结构预测、X射线晶体学解析及定点突变验证。
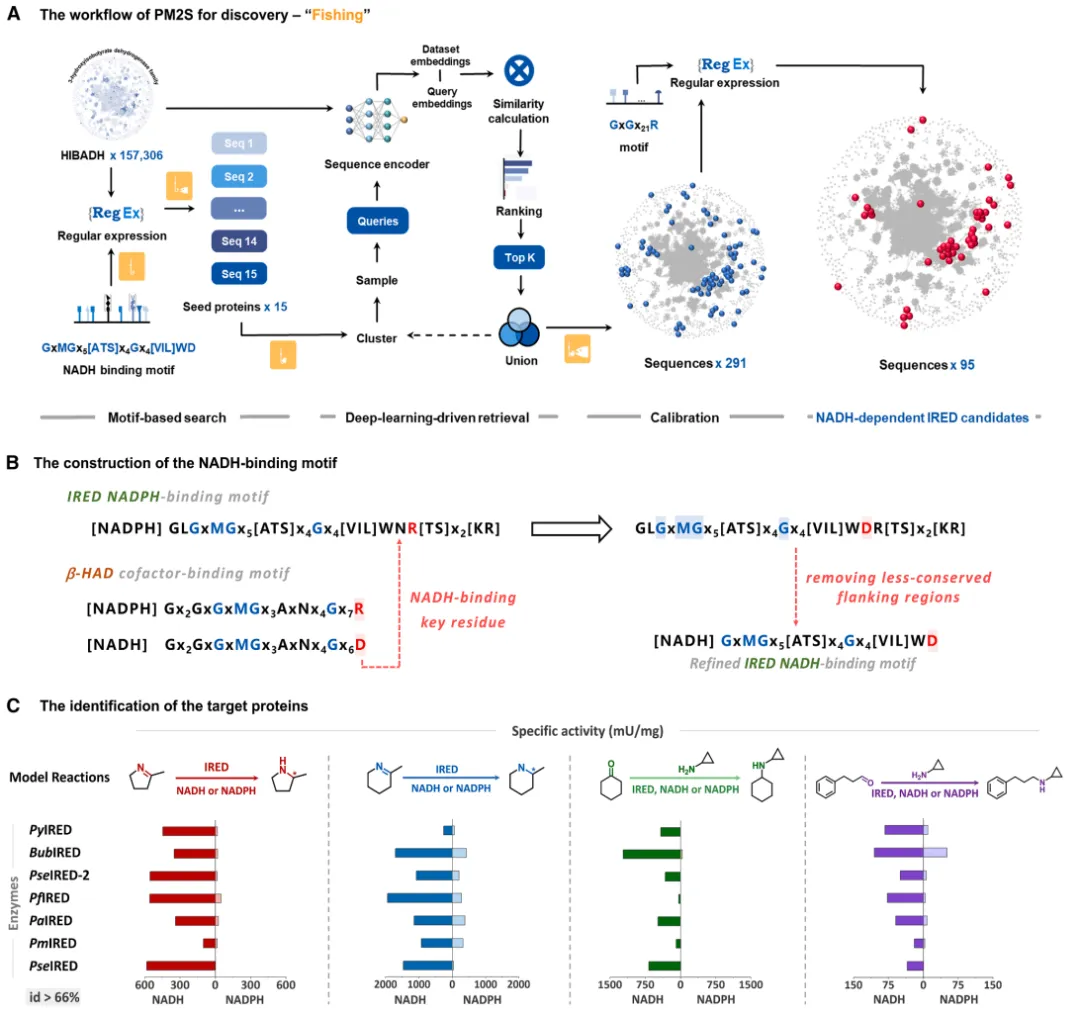
图 2. PM2S 平台挖掘出的新型 NADH 依赖型 IREDs 分析。 展示了候选蛋白与已知蛋白之间极低的序列同源性分布 (A),候选酶群的序列相似性网络 (SSN) 聚类情况 (B),代表性酶的辅因子偏好性 (C),以及新发现的结合基序的多样性 (D)。
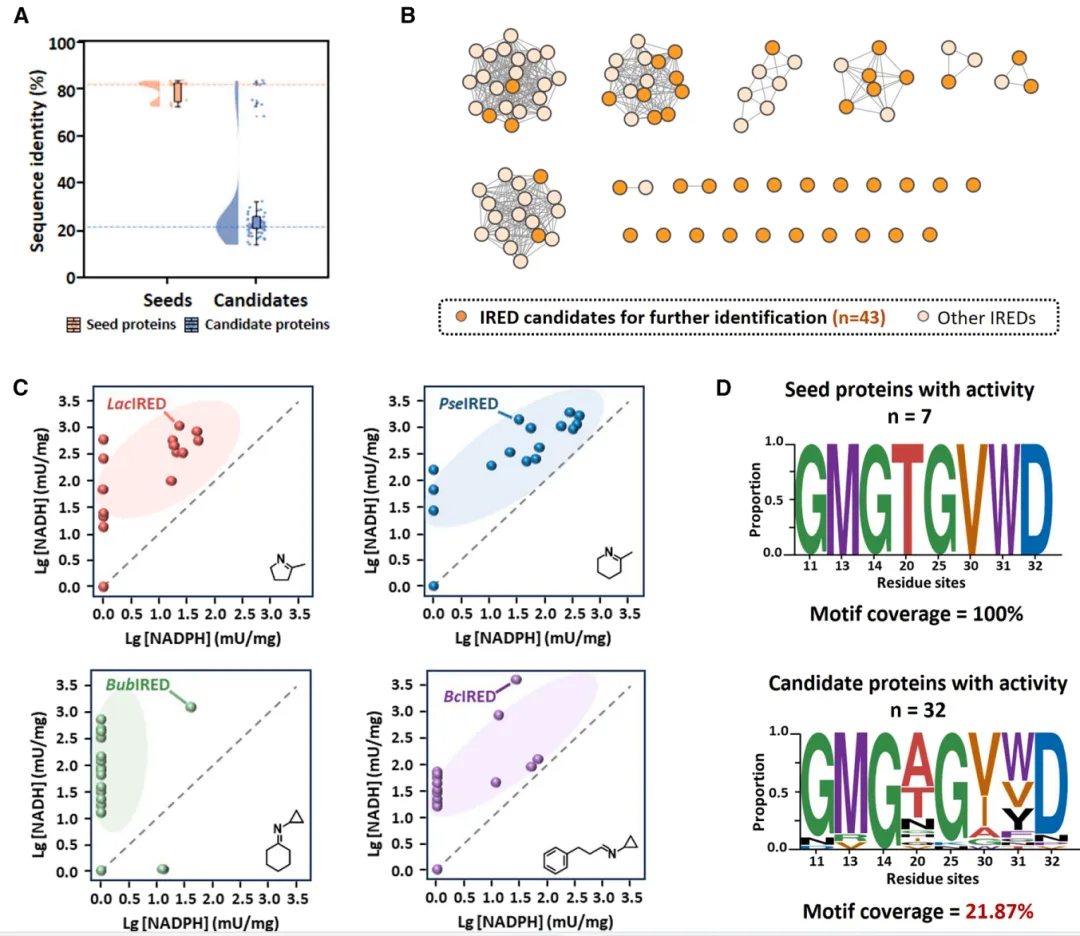
图 3. 四种代表性 IRED 催化亚胺还原 (IR) 和还原胺化 (RA) 的底物谱及活性热图比较。
10. 研究结论
成功跨越同源性鸿沟:通过 PM2S 策略,成功鉴定出95 个此前未知的 NADH 依赖型 IREDs,它们与现有已知 IREDs 的序列相似度极低(仅 12%-43%),属于一个进化上独特的全新分支。
催化性能极其优异:表征的酶展现出了对 NADH 极其显著的偏好性,并在亚胺还原(IR)和还原胺化(RA)中表现出极为宽泛的底物耐受性,甚至能够接受大位阻的萘基取代亚胺和复杂的环酮。
确定了分子特异性机制:解析了 BubIRED 与辅因子的晶体结构,证实了Asp32 是决定 NADH 特异性的核心门控残基。NADH 的核糖羟基与 Asp32 形成了紧密的氢键网络,而 NADPH 的磷酸基团会由于静电排斥被该位点排斥。
图 4. NADH 依赖型 IREDs 催化不同取代基的亚胺还原转化。
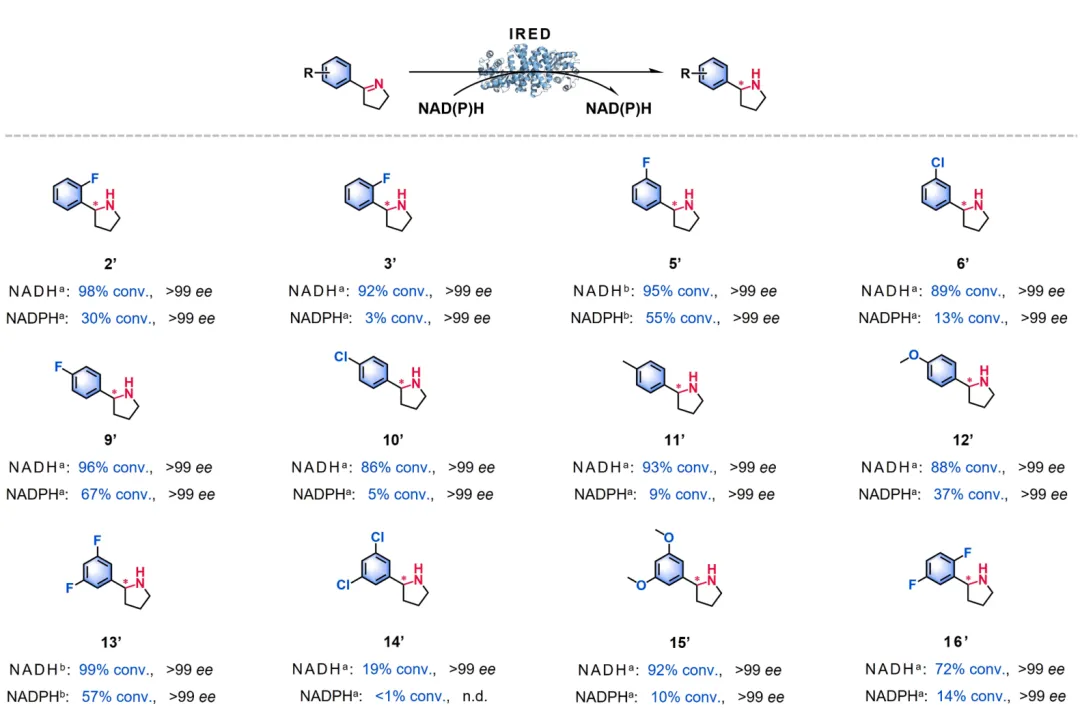
图 5. NADH 依赖型 IREDs 催化的一系列高难度醛/酮还原胺化反应。
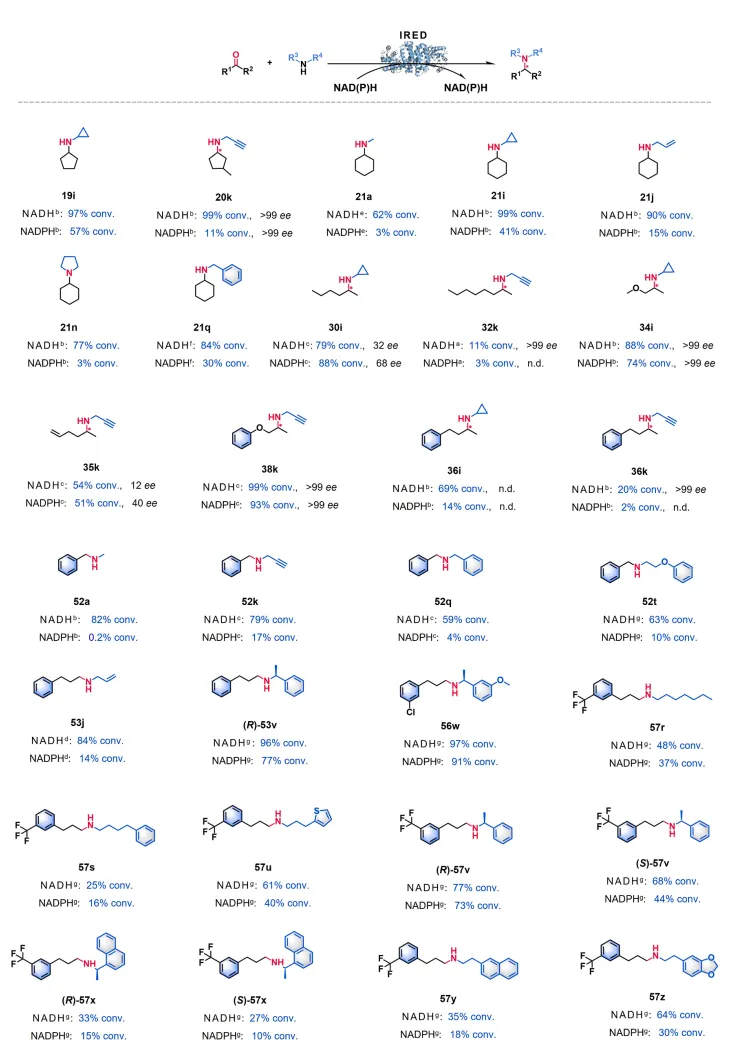
图 6. 揭示辅因子结合机制的结构分析。 (A-D) BubIRED 及其与 $\beta$-HAD 和已知 IRED 的结构比对差异;(E) 结合 AlphaFold 预测的酶-辅因子-底物三元复合物的活性口袋交互机制细节分析。
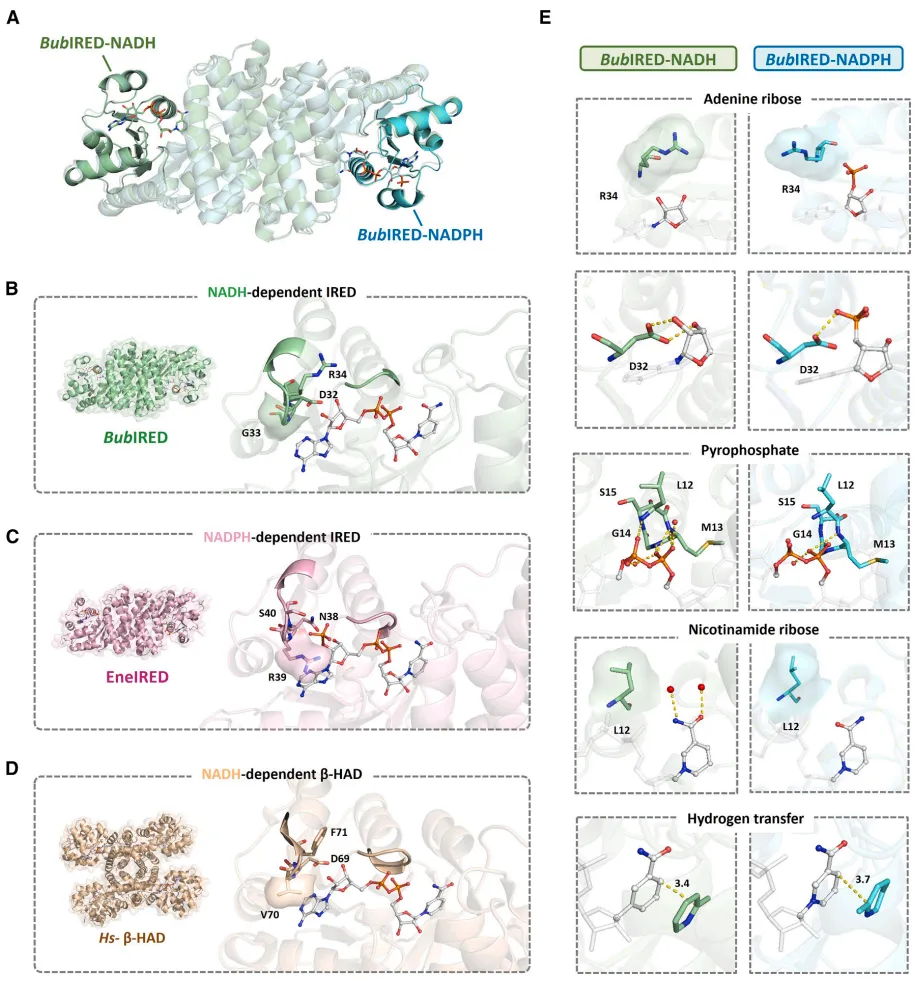
11. 研究局限性
虽然通过单点突变(Asp突变为Arg)可以在某些 NADH 依赖型酶中成功将其偏好性反转为 NADPH,但在已知的经典 NADPH 依赖型 IREDs 中进行反向突变(Arg 突变为 Asp)却失败了。这表明天然 NADPH-IREDs 的辅因子口袋可能在结构上非常刚性,仅仅依靠单点突变无法逆转其偏好,未来仍需要借助多位点组合甚至计算工具(如 CSR-SALAD)进行更深度的酶改造。
图 7. NADH 依赖型和 NADPH 依赖型 IREDs 的关键辅因子结合基序区域多序列比对。 (直观显示了32位保守的 D 与 R 残基分布规律)

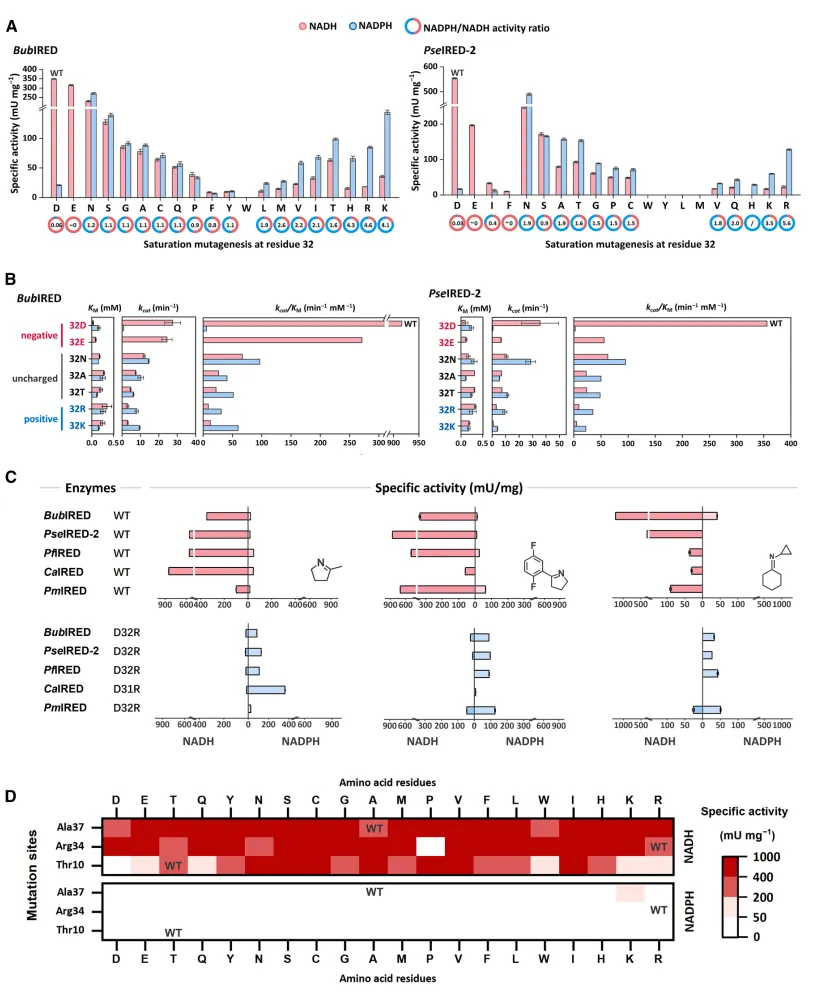
图 8. 验证关键氨基酸功能的突变分析。 (A) BubIRED 与 PseIRED-2 在32位点的饱和突变比活数据;(B) 动力学参数变化趋势验证;(C-D) 多个 IRED 候选者进行点突变后的辅因子特异性逆转确认。
12. 研究创新点
方法学创新:首次开发出将“规则匹配(Rule-based Motif)”与“AI蛋白质语言大模型(PLMs)”融合的挖掘框架(PM2S)。解决了传统方法因“序列同源性极低”导致的假阴性,以及单纯使用 AI 盲筛带来的高假阳性与高成本问题。
发现全新酶族:打破了 IRED 家族几十年来“仅限使用 NADPH”的认知,挖掘并验证了自然界首批天然存在、催化性能优异的 NADH 依赖型 IREDs。
13. 研究贡献
该工作为整个新酶挖掘领域提供了一种通用的、低成本的智能化工作流模板。挖掘出的大量新型 NADH-IREDs 拥有极高的工业应用前景,由于 NADH 更廉价且易于实现辅因子再生循环,此发现为未来制药工业中大规模、连续流动、低成本的手性胺(以及空间大位阻的药物中间体)的生物催化合成铺平了道路。
