 夜雨聆风
夜雨聆风在高通量筛选(High-Throughput Screening, HTS)驱动的药物发现早期阶段,小分子胶体聚集体(Small Colloidally Aggregating Molecules, SCAMs)是导致假阳性结果的主要因素之一。据估计,公共化学基因组数据库中约15%–20%的小分子在标准筛选条件下会发生聚集,这些聚集体通过非特异性结合或聚集诱导发光等机制干扰靶点功能,严重降低了筛选数据的可靠性。虽然动态光散射等实验方法可检测聚集,但成本高且难以预测。现有的计算工具(如Aggregator Advisor)多基于规则或传统机器学习(如梯度提升树),存在准确率有限(约70%–80%)、无法规模化筛选大库或缺乏可解释性等瓶颈。
因此,加拿大曼尼托巴大学化学系 Rebecca L. Davis教授与德国卡尔斯鲁厄理工学院Pascal Friederich教授摒弃了传统的“黑箱”模型思路,采用可解释人工智能(Explainable AI, xAI)框架,构建了多通道图注意力网络(Multi-channel Explanation Graph Attention Network, MEGAN),开发了一种兼具高精度、可扩展性及结构洞察力的新方法,以加速先导化合物的优化。该工作以“Mitigating Molecular Aggregation in Drug Discovery With Predictive Insights From Explainable AI”为题发表在《Angewandte Chemie International Edition》期刊上。

该工作主要将分子表示为图结构(原子为节点,化学键为边),利用图注意力机制自动学习分子子结构对聚集行为的贡献。模型不仅输出二分类结果(聚集/非聚集),更通过内置的注意力掩码(Attention Mask)机制,为每个预测生成可视化的归因解释(Attributional Explanation),直观展示促进聚集(橙色)或抑制聚集(绿色)的关键药效团。此外,模型集成了反事实解释(Counterfactual Explanation)功能,能够针对查询分子自动生成结构相似但预测标签相反的衍生物,为药物化学家提供具体的结构修饰建议,实现从“预测问题”到“设计解决方案”的闭环。
创新点
1. 可解释图神经网络的精准预测:首次将MEGAN模型应用于SCAMs预测,在包含18万+分子的数据集上达到82%的准确率与81%的F1分数,性能优于传统的ChemAgg(XGBoost)模型。其创新在于将解释生成过程内置于模型前向传播中,而非事后分析,保证了解释与预测的逻辑一致性。
2. 反事实引导的分子设计:突破了传统分类器仅提供“是/否”判断的限制,开发了配套的反事实生成器。用户输入一个聚集分子,模型可快速生成结构微调(如官能团替换)后的非聚集类似物,并通过氯碘羟喹(Clioquinol)衍生物的合成与实验验证,证实了该策略的实用性。
3. 物理机制关联与全局概念挖掘:通过密度泛函理论(Density Functional Theory, DFT)计算证实,模型高置信度预测为聚集体的分子(如2-羟基吡啶)其分子间相互作用能(氢键、π–π堆积)显著更强,验证了模型敏感性的物理基础。同时,通过全局概念提取(Global Concept Extraction)技术,首次系统性地揭示了尿素基团(Urea)与聚集正相关、硫脲(Thiourea)与聚集负相关(除非邻位有吸电子基团)等非直观化学规律。
图文信息
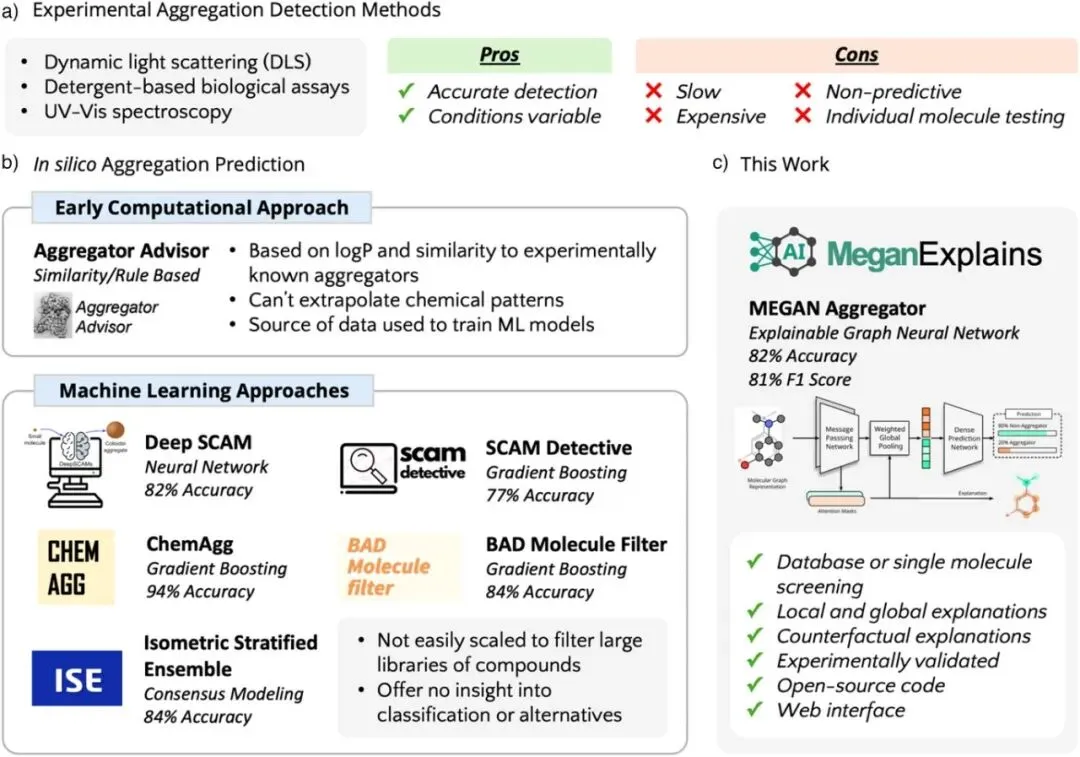
图1:实验与计算聚集预测方法的对比,展示了药物发现中应对小分子胶体聚集(SCAMs)的演进路径。a) 部分列举了动态光散射(DLS)、去污剂生物测定和UV-Vis光谱等实验检测手段,指出其虽然准确,但存在速度慢、昂贵且难以大规模筛选的缺陷。b) 部分总结了早期计算方法(如基于logP的Aggregator Advisor)及后续各类机器学习(ML)方法(如Deep SCAM、ChemAgg等),指出传统计算方法无法外推化学模式,现有ML模型往往缺乏开源实现或难以解释。c) 部分展示了本研究提出的MEGAN模型,它整合了图神经网络(GNN)的预测能力与可解释AI(XAI)技术,支持数据库或单分子筛选、局部/全局解释、反事实解释,并已通过实验验证。
确立了MEGAN模型在药物发现背景下的优势地位,即通过结合高准确度的预测与XAI技术,填补了现有计算方法在可解释性、可扩展性及提供结构修改建议方面的技术空白 。为药物化学家提供了一个能够直接指导化合物优化、减少高通量筛选中假阳性的有力工具。
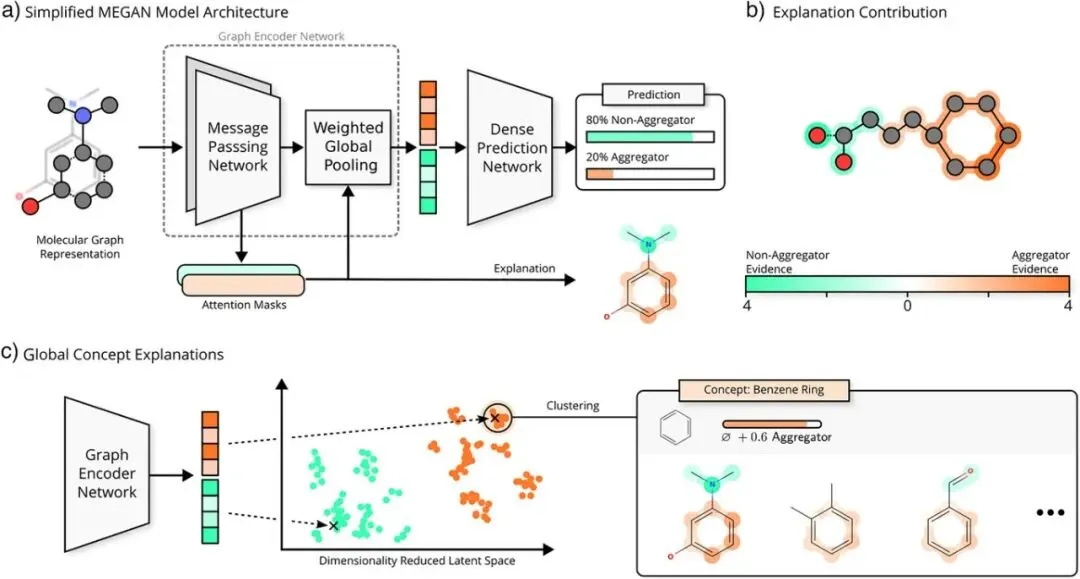
图2:MEGAN模型架构与可解释性工作流程,a)展示了简化的MEGAN模型架构流程:分子图结构被输入到多个基于注意力的消息传递层,节点表征被聚合后传递到全连接网络以输出预测类别,解释掩码则从内部的注意力值中导出。b)说明了基于注意力的解释如何被明确地分割为对应每个可能输出类别(如聚集剂或非聚集剂)的独立通道。例如,橙色通道突出显示支持“聚集剂”分类的结构证据,而绿色通道则突出显示支持“非聚集剂”分类的结构证据。c)描述了如何通过分子子图嵌入的潜在空间进行聚类来发现更高层级的结构解释,分析同一概念簇中的所有成员可以得出与某些结构模式相关的普遍趋势。图中明确指出,为了便于说明,解释掩码和值是构建出来的。
MEGAN模型的核心创新架构设计在于,将“解释生成”与“预测推断”紧密结合在同一网络前向传播过程中。它不是“事后诸葛亮”式的分析,而是通过多通道注意力机制,在预测的同时,为每个类别(聚集/非聚集)生成直观的、可视化的原子/键层面归因图。此外,通过全局概念提取技术,可以系统性地挖掘隐藏在模型决策背后、与特定分子亚结构相关的化学模式,实现了从“单分子解释”到“规律性发现”的跃升。
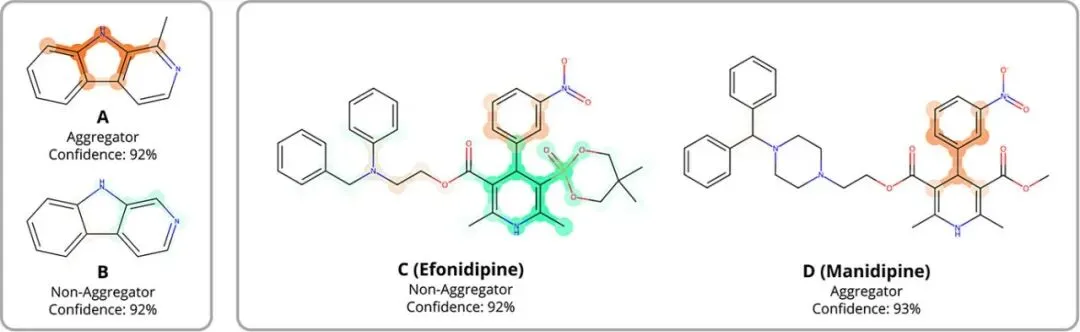
图3:结构敏感性预测示例。通过两组分子对(A/B和C/D),展示了MEGAN模型对细微结构修饰的敏感性及其解释能力。第一组(A, B):氮杂咔唑A(被文献报道为聚集剂)与B(非聚集剂)仅相差一个甲基基团。MEGAN模型不仅正确预测了两者的类别,其解释掩码还显示,这个甲基的存在导致模型关注了两种分子共有的杂环核心的不同部分来进行分类,这表明甲基的引入改变了模型对核心结构“化学环境”的解读,而非甲基本身被直接视为关键特征。第二组(C, D):非洛地平(C, 非聚集剂)与马尼地平(D, 聚集剂)。模型在两者中都识别出硝基苄基的芳香环是支持聚集的证据。在C中,庞大的膦酸酯基团被识别为反对聚集的证据,这从化学角度是合理的,因为其位阻可能抑制紧密堆积。而在D中,甲酯基团的存在削弱了二氢吡啶环部分的“反聚集”归因,暗示甲酯对促进聚集的分子间相互作用的干扰较小。
MEGAN模型展现出对“细微结构变化导致聚集行为改变”这一药物化学核心议题的深刻捕捉能力。其解释掩码不仅能验证化学直觉(如庞大基团抑制聚集),更能揭示反直觉的案例(如单个甲基导致聚集行为翻转),并指出结构变化如何影响模型对分子其他部分的“感知”。这证明MEGAN不仅能做出准确预测,更能提供超越简单指纹特征的、基于化学环境的、可解释的结构活性关系洞察,有助于研究者理解结构修饰的具体影响机制。
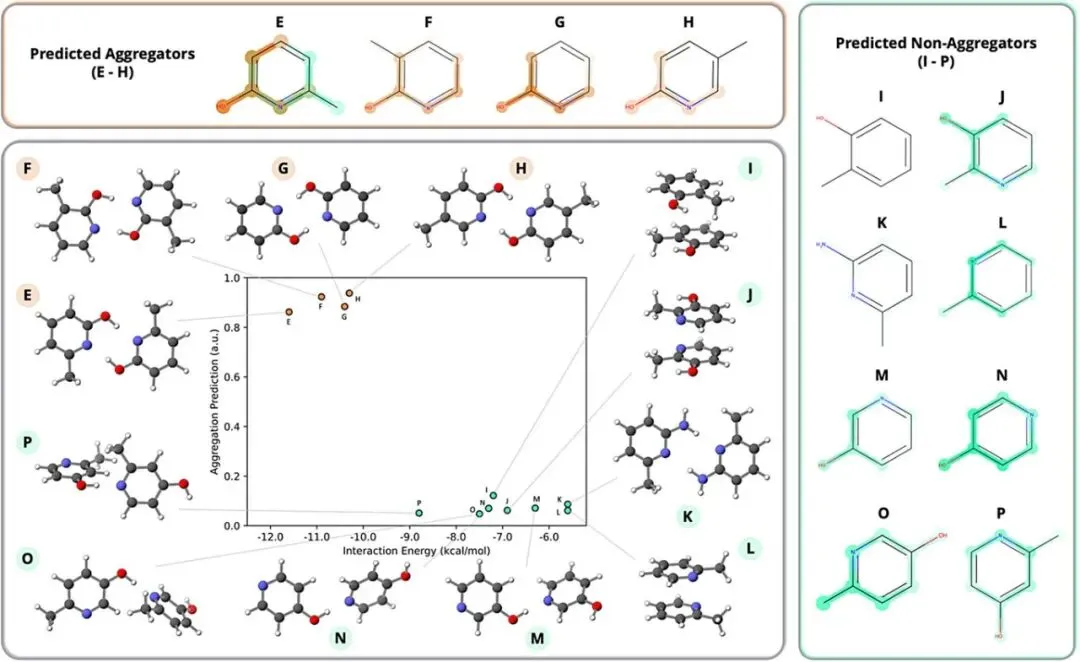
图4:MEGAN预测与密度泛函理论(DFT)相互作用能的对比。选取了一组吡啶衍生物(E-P),将MEGAN的聚合倾向预测值与通过DFT计算得出的二聚体相互作用能进行关联。结果显示,高置信度的聚合分子(E-H)表现出更强的分子间相互作用能(更负),且模型通过解释掩码成功锁定了HO-C-N这一氢键供体基团。对于被分类为非聚合的异构体,模型则识别出其羟基的特定取代模式导致了较弱的相互作用。
该图有力证明了MEGAN的预测结果与基于量子力学的物理化学相互作用具有高度一致性,其解释机制能够从分子间相互作用强度的角度提供符合物理直觉的科学依据 。
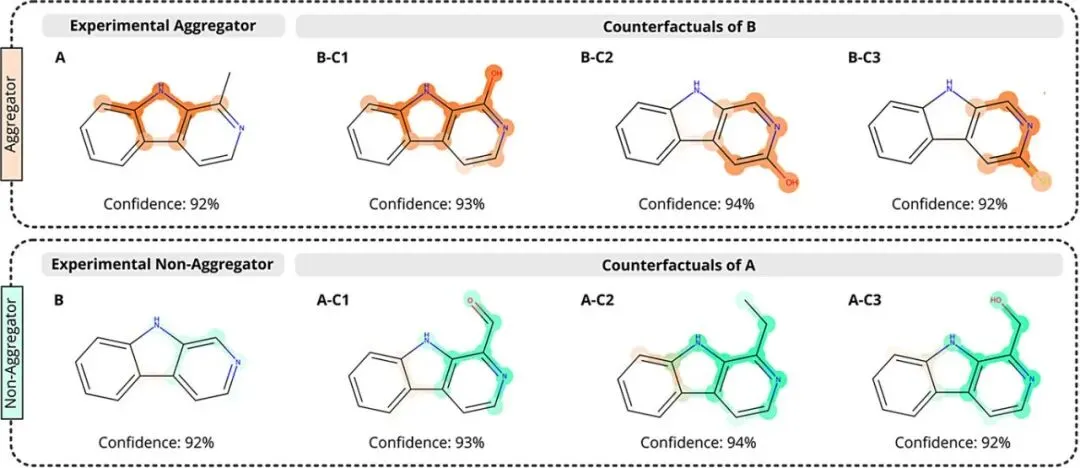
图5:针对分子的反事实解释生成。演示了通过反事实(Counterfactuals)分析来探测结构与聚合属性的逻辑关系。以聚合分子A为例,通过引入醛基(A-C1)或增大位阻,模型将其预测结果“逆转”为非聚合;反之,在非聚合分子B的pyridine氮原子邻位引入羟基或硫醇(B-C1/C2/C3)则诱导了聚合趋势。
展示了MEGAN不仅能分类,还能通过生成结构高度相似但标签相反的“反事实”分子,为药物化学家提供直接的结构优化方案,以降低先导化合物的聚合风险 。
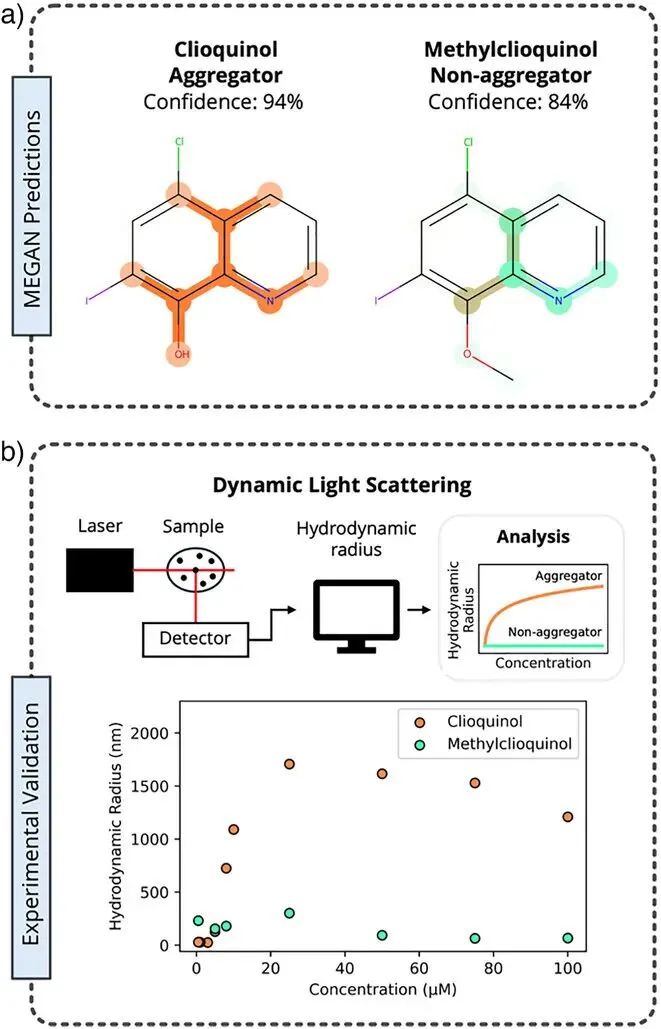
图6:Clioquinol及其衍生物的实验验证。通过动态光散射(DLS)实验对模型建议的修改方案进行了最终验证。Clioquinol被证实具有聚合性,而模型预测其甲基化衍生物(Methylclioquinol)为非聚合分子。实验数据显示,Clioquinol在浓度增加时水力学半径显著增大,而Methylclioquinol则保持稳定。a部分显示,模型通过解释掩码识别出Clioquinol氮氧结构参与的氢键是聚合关键,而甲基化阻断了这一互动。
这是对MEGAN预测能力与反事实设计逻辑的最直接实验支撑。它证明了MEGAN生成的结构建议在实验室条件下是真实有效的,能够通过微小的化学修饰实现化合物物理行为的理性控制 。
详细信息请点击左下角“阅读全文”
H. Sturm, J. Teufel, K. A. Isfeld, et al., Mitigating Molecular Aggregation in Drug Discovery With Predictive Insights From Explainable AI. Angewandte Chemie International Edition 2025, 64, e202503259.
