 夜雨聆风
夜雨聆风

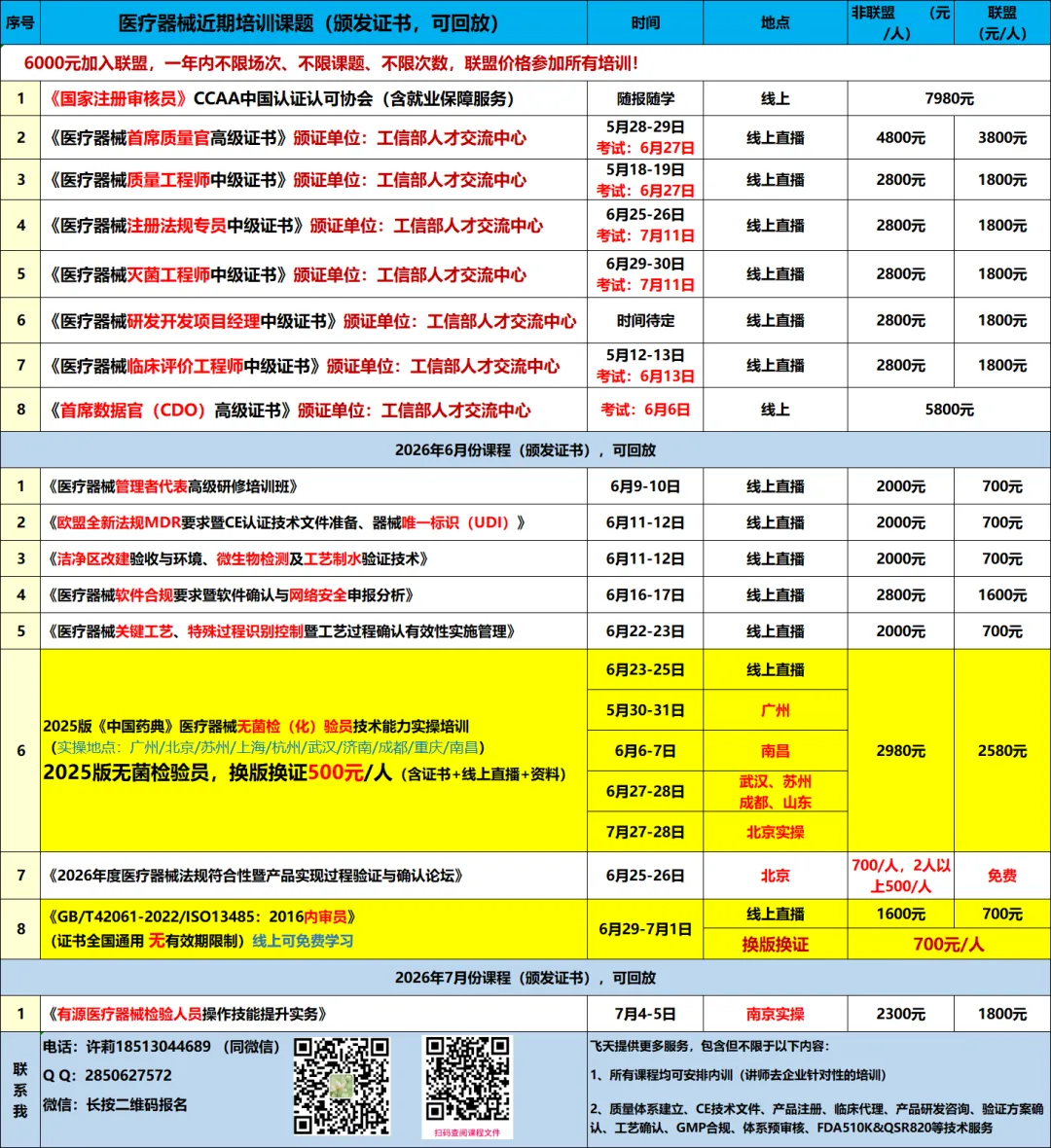
《医疗器械生产质量管理规范》实施以来,国家监管部门相继发布了无菌医疗器械、植入性医疗器械、体外诊断试剂等多个附录文件,持续完善生产质量监管体系。2019年7月,国家药监局发布《医疗器械生产质量管理规范附录独立软件》,进一步构建了规范独立软件生产质量管理的技术文件体系。2022年3月,国家药监局器审中心又修订并发布《医疗器械软件注册技术审查指导原则》,为医疗器械软件的生存周期过程管理及注册申报提供了明确指导,也为体系核查工作提供了重要依据。
随着软件在医疗器械中的作用日益关键,其可靠性已成为行业关注的核心。软件一旦发生运行失效,可能导致设备故障、诊断延误甚至错误治疗,给患者带来严重伤害乃至生命危险。为助力企业深入理解合规要求,有效开展软件确认工作,确保软件持续符合预期用途,并应对网络安全申报的挑战,飞天教育特组织本次专题会议。现诚邀贵单位的积极参与,现将有关事项通知如下:

1、医疗器械软件注册审查指导原则解析;
2、欧盟MDCG & FDA 510k软件文档要求介绍;
3、YY/T 0664-2020医疗器械软件 软件生存周期过程技术要求;
4、软件生存周期中的风险管理与重点(结合GB/T42062 相关标准);
5、软件产品设计开发流程说明与软件/系统测试验证及确认要求的有效性实施;
6、软件产品注册申报中“器械安全有效基本要求清单”分析与申报资料的完整性;
7、医疗器械设备软件描述文档编写规范;
8、医疗器械网络安全基本概念;
9、医疗器械网络安全注册审查要求;
10、医疗器械网络安全验证与确认的相关质控要求与实施;
11、医疗器械网络安全生存周期;
12、医疗器械网络安全风险考虑与风险管理活动;
13、医疗器械网络安全风险考虑与风险管理活动上市后管理;
14、讨论与答疑。

医疗器械监管机构、技术支持机构等相关工作人员;医疗器械企业从事生产、研发、注册的人员、技术部长、注册文件编写人员等;医疗器械企业负责验证和确认工作的人员等。

参会证书:学员需全程参与课程学习,学习结束后将获得“医疗器械软件合规要求暨软件确认与网络安全申报分析”培训证书。


 长期举办 管代、13485内审员、微生物无菌检验员等
长期举办 管代、13485内审员、微生物无菌检验员等
 更多培训计划或企业辅导请联系许莉18513044689(同微信)
更多培训计划或企业辅导请联系许莉18513044689(同微信)
