 夜雨聆风
夜雨聆风
点击蓝字 关注我们


如果说 Biomni 是一位 AI 生物学家,那么 genomics.py 无疑是它最核心的大脑之一。
在整个 Biomni 系统中,基因组学与单细胞分析模块承担着大量高价值科研任务:从单细胞数据分析、细胞类型注释,到染色质互作分析、跨物种基因转换,再到蛋白语言模型嵌入生成,几乎覆盖了现代生物信息学研究的核心流程。
官方文档显示,整个 genomics 工具库共集成了 21 个专业函数,并按照功能划分为四大模块:单细胞分析、基因分析、比较基因组学以及表观遗传学分析。
Biomni 的单细胞工具究竟有多强?
当前,单细胞分析已经成为生命科学研究中最热门的方向之一。
而在 Biomni 中,单细胞分析并不是一个独立工具,而是一整套完整的分析工作流。官方工具库提供了 7 个核心函数,可以完成从原始表达矩阵到最终细胞注释的全部流程。
整个流程大致如下:
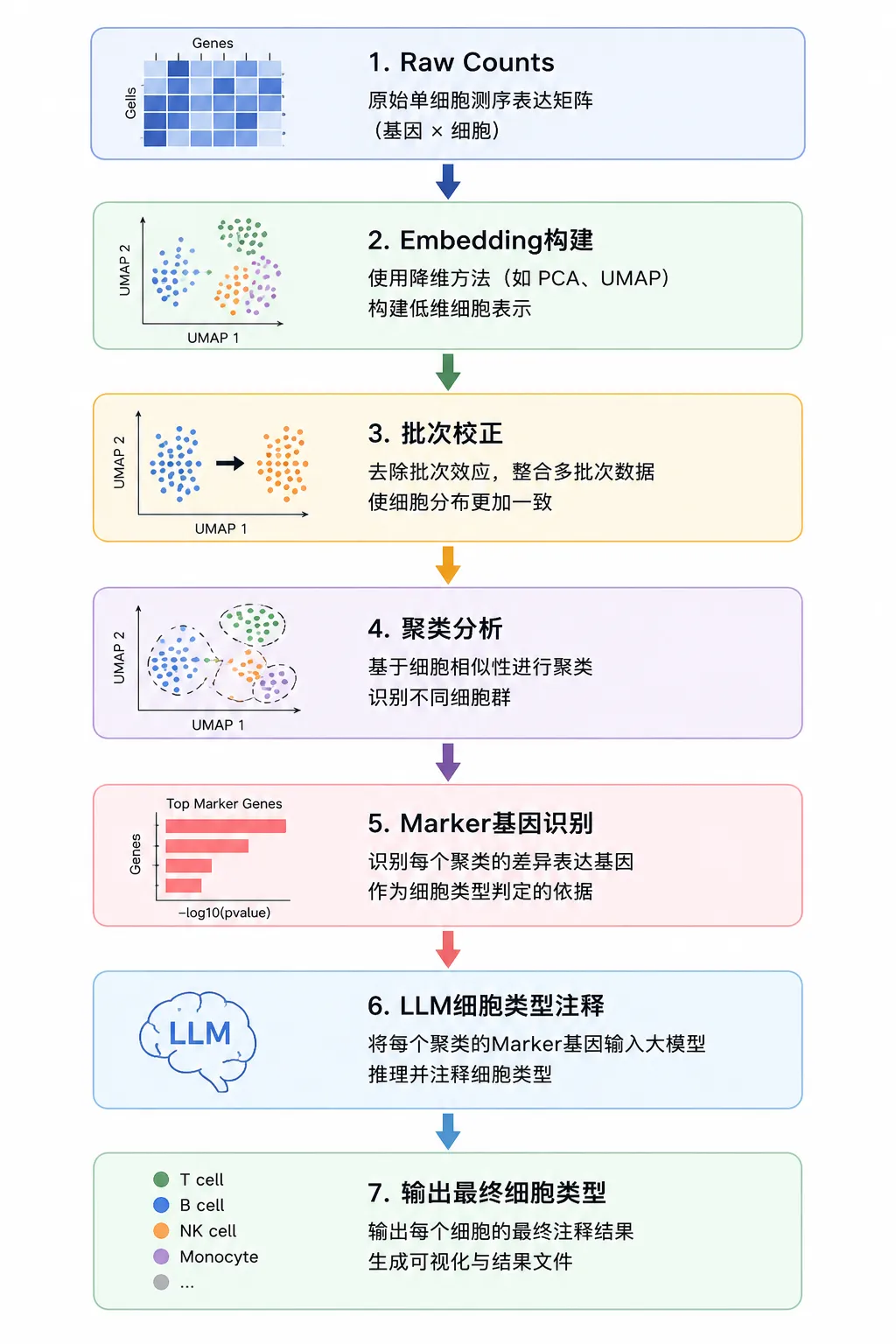
与传统分析流程最大的不同在于:
Biomni 将大模型真正引入到了单细胞注释环节。
LLM如何给细胞“命名”?
在传统单细胞分析中,研究人员通常需要根据 Marker Gene 手动查阅文献,进而判断细胞类型。
而 Biomni 中的:
annotate_celltype_scRNA()函数则尝试让大模型直接完成这一过程。
其工作流程大致如下:
提取每个 Cluster 的差异表达基因;
将 Marker Gene 输入大模型;
大模型推理可能的细胞类型;
与 CZI Cell Census 细胞本体数据库进行校验;
如果预测结果不合法,则重新推理。
最终,大模型需要按照如下格式输出:
Cell Type ; Confidence Score ; Reason例如:
CD4+ T cell ; 0.93 ; Expresses IL7R, LTB and MAL随后,Biomni 会自动将:
cell_typecell_type_reason
写入 AnnData 对象中,形成最终注释结果。
换句话说,过去需要人工查阅大量文献的步骤,现在已经能够由 AI 自动完成。
除了大模型,Biomni 还集成了哪些细胞注释方法?
事实上,Biomni 并没有完全依赖大模型。
为了保证注释的准确性,系统同时集成了多个主流单细胞注释框架。
1.Azimuth 神经网络注释对应函数:
annotate_celltype_with_panhumanpy()该方法基于 Azimuth 神经网络模型,可以完成跨组织、跨数据集的层级化细胞注释。
由于依赖环境复杂,开发团队甚至专门为其创建了独立 Conda 环境,并通过子进程的方式调用。
这也体现出 Biomni 的设计理念:
与其重新实现算法,不如直接调度最优秀的软件。
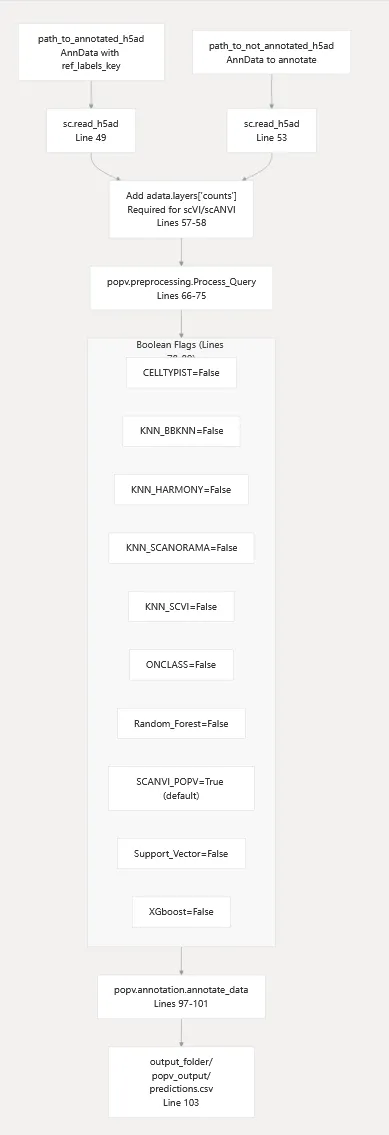
2.popV 集成注释框架对应函数:
unsupervised_celltype_transfer_between_scRNA_datasets()该工具基于 popV 实现参考数据集标签迁移。
最令人惊讶的是:
它一次性集成了 10 种分类器,包括:
SCANVI
KNN
Random Forest
XGBoost
多种深度学习模型
用户甚至可以同时开启多个模型,并通过集成学习获得最终预测结果。
Embedding:Biomni 如何理解单个细胞?
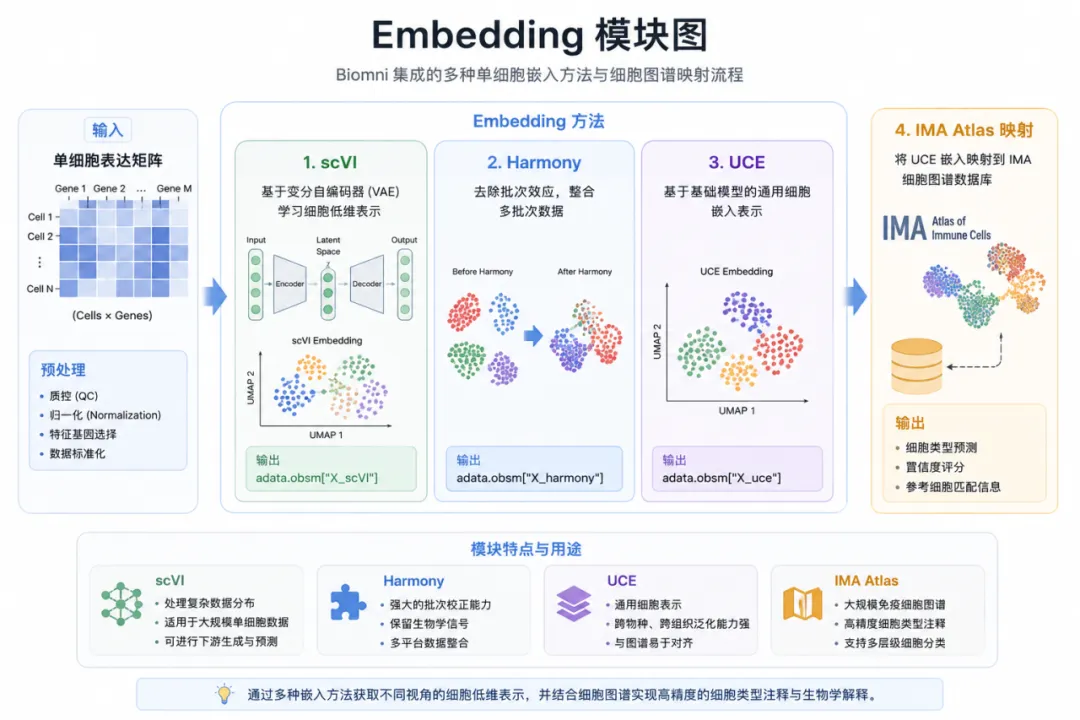
在现代单细胞分析中,Embedding 已经成为核心技术。
Biomni 同样集成了多种主流嵌入模型:
scVI通过变分自编码器学习细胞低维表示输出:
adata.obsm["X_scVI"]Harmony用于去除批次效应输出:
adata.obsm["X_harmony"]UCE(Universal Cell Embedding)利用基础模型生成通用细胞表示输出:
adata.obsm["X_uce"]在生成 UCE Embedding 后,Biomni 还能够进一步映射至 IMA 细胞图谱数据库,实现自动细胞解释。
genomics.py 不只是单细胞分析
除了单细胞分析外,该模块还集成了大量高级基因组学工具。
跨物种基因转换函数:
interspecies_gene_conversion()支持人、小鼠、斑马鱼等 13 个物种之间的基因同源映射。底层通过 Ensembl BioMart API 实现。
基因集富集分析函数:
gene_set_enrichment_analysis()底层调用:
gget.enrichr支持:
KEGG
GO
ChEA
Reactome
等多个数据库。
蛋白语言模型函数:
generate_gene_embeddings_with_ESM_models()利用 Meta 开源的 ESM 模型生成蛋白序列嵌入表示。
甚至支持数十亿参数规模的大模型。
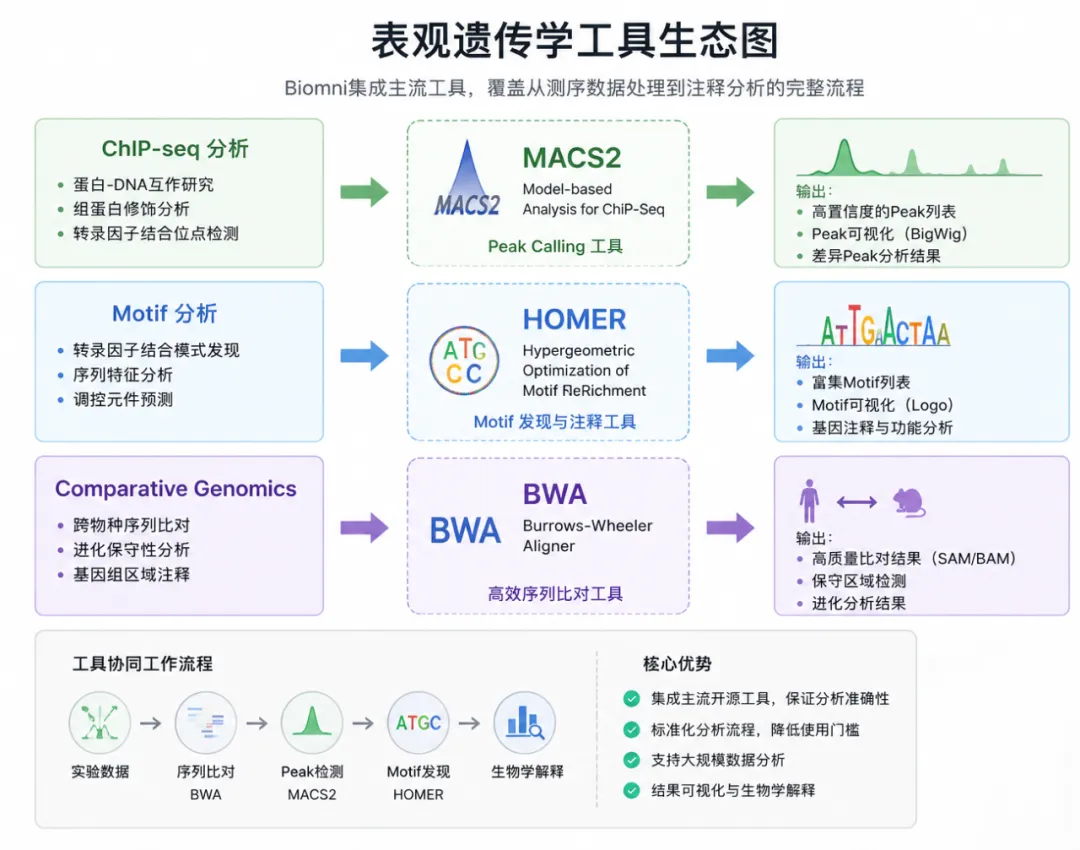
染色质与表观遗传学分析
除了转录组,Biomni 同样支持表观遗传学研究。
主要功能包括:
ChIP-seq Peak Calling
染色质互作分析
Motif 富集分析
基因组区域重叠分析
系统直接调用:
MACS2
HOMER
BWA
等经典生信软件完成分析。
这意味着:
Biomni 本质上并不是一个新的算法框架。
而是一个能够自主调用顶级生信工具的大模型智能体。
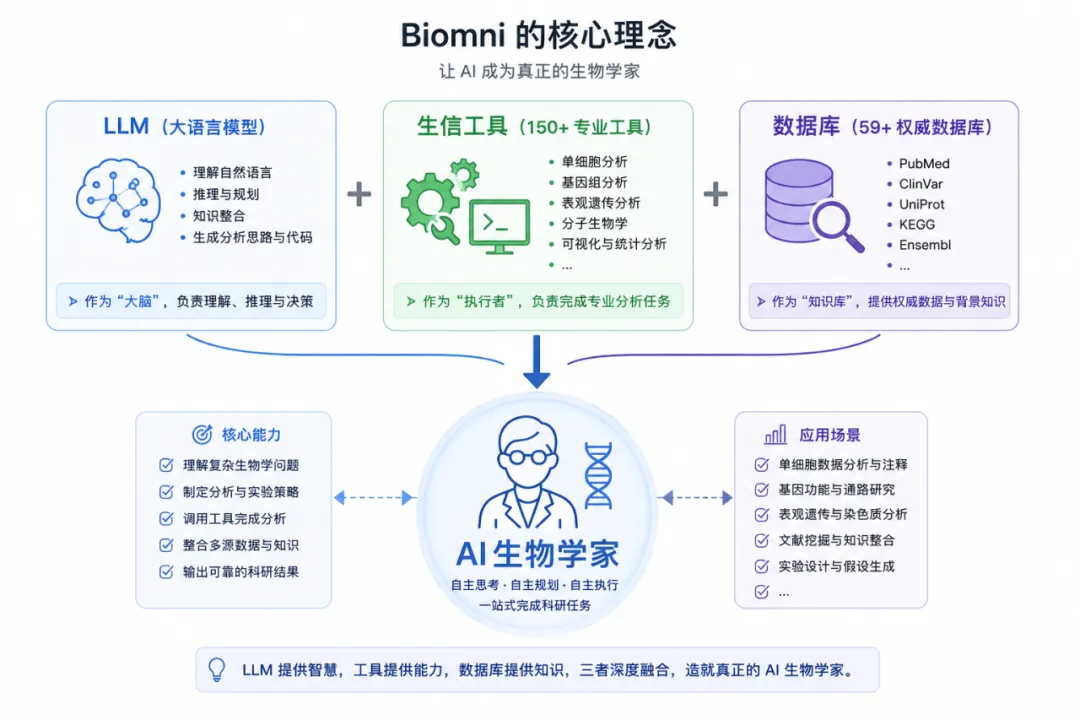
阅读完整个 genomics.py 模块后,我最大的感受是:
Biomni 并没有重新发明生信算法。
相反,它选择了一条更现实的路线:
利用大模型作为“大脑”,调度 Scanpy、scVI、MACS2、HOMER、ESM 等成熟工具,最终构建出一个真正能够执行科研任务的 AI 生物学家。
其实这种设计思路,在科研工具里是共通的。
比如在 青熵视界(https://qssj.nextsci.cn)
这类数据分析与可视化系统里,也在做类似的事情:
用结构化流程替代手工分析
用参数化系统替代经验选图
用规则约束保证图表一致性
用自动化生成降低科研表达成本
本质上两者在解决同一个问题:
把“经验驱动的科研操作”,变成“可计算、可复现、可约束的系统”。
Biomni 在做的是“AI 生信自动注释系统”,
而青熵这类工具在做的是“科研表达与分析自动化系统”。
方向不同,但底层逻辑一致:
让科研从“人做流程”,变成“系统执行认知”。
点击
阅读原文
查看更多
