 夜雨聆风
夜雨聆风





重磅推荐!AI蛋白质设计与抗体设计与AI基因编辑线上直播!
01 AI基因编辑线上直播课
02 AI辅助抗体设计线上直播课
03 AI蛋白质设计线上直播课
04 合成生物学与基因线路设计线上直播课
05 深度学习在多组学融合中应用线上直播课
06 机器学习代谢组学线上直播课
优惠1:报二送一(同时报名两个班赠送一个学习班,赠送班任选)
优惠2:提前报名缴费可享受300元优惠(仅限前15名)
优惠3:报名直播课程可赠送往期课程回放

主讲老师在学术界具有多年的研究经历和应用经验,来自于国内顶尖课题组,从事基因组编辑技术与人工智能交叉融合的研究工作,相关工作成果已在Nature Biotechnology、Nature Plants、Trends in Biotechnology等国际知名期刊发表
第一天
1. 基因组编辑技术简述
1.1 基因组测序、编辑和读写时代及基因组编辑技术现状简述
2. 基因组编辑四代技术原理
2.1 四代基因组编辑技术发展历程
2.2 ZFN、TALEN和CRISPR/Cas系统的组成和工作原理
3. CRISPR/Cas系统的来源及分类
3.1 CRISPR/Cas系统的发现过程
3.2 CRISPR/Cas系统的适应性免疫原理
3.3 CRISPR/Cas系统的分类依据和类型
4. CRISPR/Cas系统介导的DNA编辑工具
4.1 CRISPR/Cas9基因编辑工具
4.2 CRISPR/Cas12a基因编辑工具
5. CRISPR/Cas系统衍生工具的发展
5.1 碱基编辑工具的组成、作用原理及其应用
5.2 引导编辑的作用机理、应用及其发展动态
6. CRISPR/Cas介导的基因调控、细胞成像和核酸检测技术
6.1 CRISPR/Cas介导基因调控技术的原理和工具组成
6.2 CRISPR/Cas介导细胞成像技术的原理和工具组成
6.3 CRISPR/Cas介导核酸检测技术的原理和工具组成
第二天
1. 脱靶效应及其检测
1.1 脱靶效应的检测方法:扩增子测序、全基因组测序、GUIDE-seq等
1.2 脱靶效应的规避方法
2. 基因编辑流程-以植物为例
2.1 靶位点sgRNA或crRNA的设计原则
2.2 表达盒设计和构建的方法
2.3 植物原生质体瞬时表达系统
2.4 基因编辑载体的遗传转化
2.5 基因编辑突变体的检测
3. 基因组编辑常用软件实操
3.1 靶位点设计软件Cas-Designer、BE-Designer、PE-Designer等
3.2 突变分析软件Cas- Analyzer、BE-Analyzer、PE- Analyzer
4. 基因组编辑技术在各领域的应用现状及前景
4.1 基因组编辑技术在基因治疗、免疫学、病毒诊断等方面的应用
第三天理论部分(人工智能+基因编辑背景)
1.深度学习概述
1.1. 深度学习的基础
1.2. 深度神经元网络的工作原理
1.3. 深度学习技术的发展趋势:自监督学习、迁移学习和少样本学习的进展
2.深度学习在基因编辑中的应用
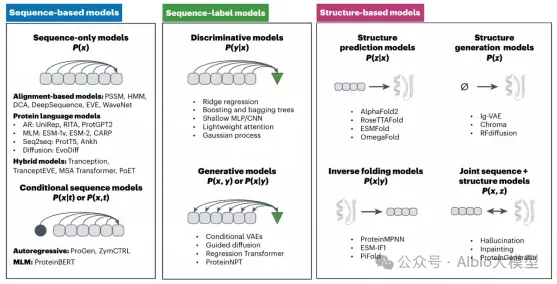
2.1. 基于监督学习的应用:序列标签模型
2.2. 零样本预测模型的应用:结构模型、大语言模型、多模态模型、
2.3. 少样本预测框架的应用(Design-Build-Test-Learn和Lab-in-the-loop范式)
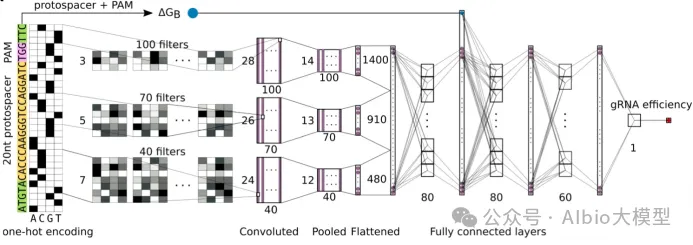
3.深度学习在gRNA优化与设计中的应用
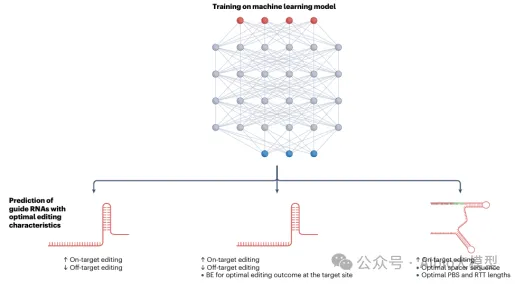
3.1. gRNA活性预测
3.2. 脱靶效应预测
3.3. gRNA预测模型介绍
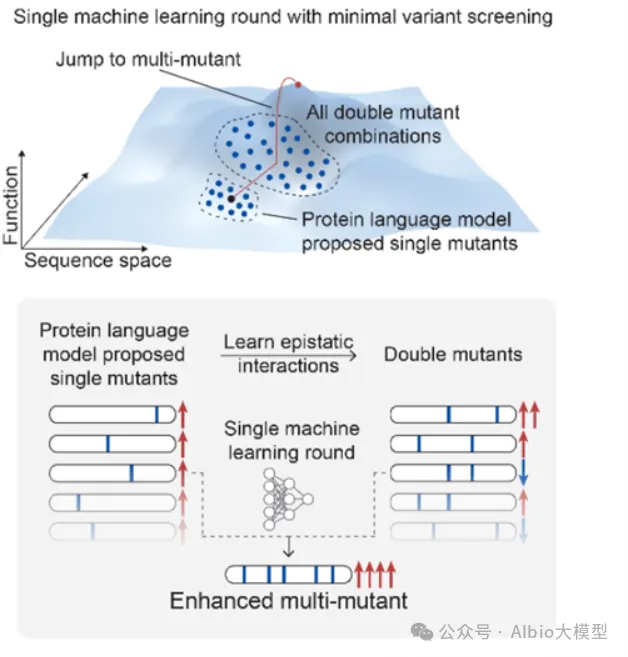
4. AI辅助的蛋白定向进化在基因编辑中的应用
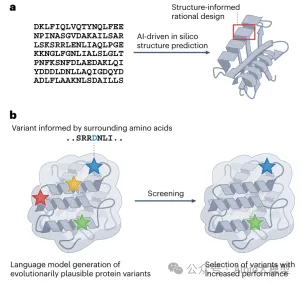
4.1. 蛋白定向进化的基本概念与实验方法
4.2 AI辅助的蛋白进化工具
4.3. AI与实验反馈的结合
5. AI蛋白质设计在基因编辑中的应用
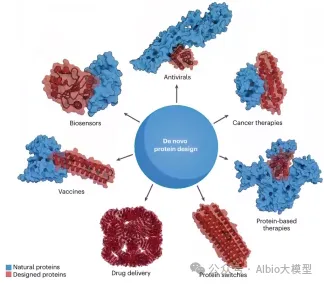
5.1. 蛋白质设计工具
5.2. 酶设计
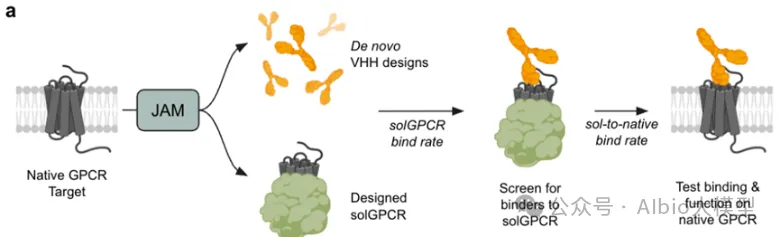
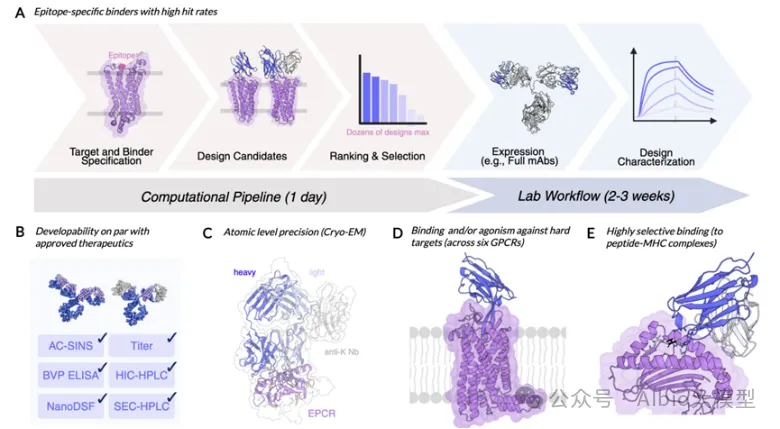
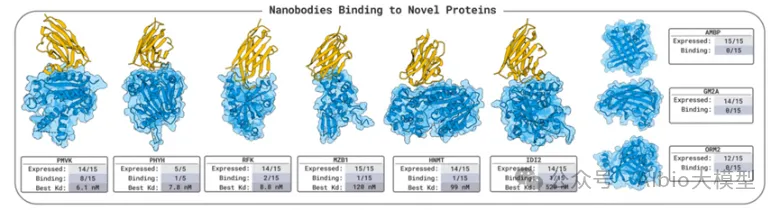
5.3. binder设计
6.AI酶挖掘在基因编辑中的应用
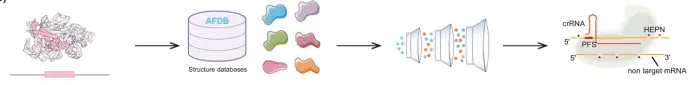
6.1. 基于大语言模型挖掘基因编辑酶
6.2. 基于结构比对挖掘基因编辑酶
第四天深度学习在基因编辑中的应用实操教学
1. 基础知识和环境搭建
1.1. GPU服务器登录
1.2. Linux基础知识
1.3. Python基础知识
1.4. 常用深度学习工具包介绍及安装
2.利用深度学习预测gRNA活性
2.1. 配置深度学习环境,安装gRNA活性预测所需的工具
2.2. 高通量数据获取:公开数据集的介绍与使用
2.3. 数据集划分:训练集、验证集、测试集
2.4. 模型搭建与调试:深度学习模型架构设计(如CNN, RNN)
2.5. 模型性能评估:精度、召回率、F1分数等评估指标
2.6. gRNA活性预测:实际应用案例演示和预测结果的解读与应用
3.利用深度学习预测编辑活性
3.1. 环境配置:安装所需工具与库
3.2. 数据获取:编辑活性相关数据集清洗
3.3. 数据集划分
3.4. 模型搭建与调试
3.5. 模型性能评估
3.6. 编辑活性预测:预测结果的展示与解读
4.零样本蛋白进化工具AiCE实操
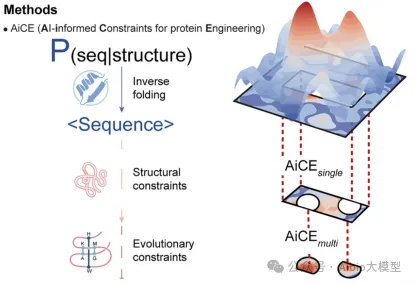
4.1. AiCE的原理与应用场景
4.2. 环境搭建
4.3. 逆折叠模型的使用:如何利用AiCE进行高活性突变预测;案例演示与实际操作
4.4. 应用实例:碱基编辑器的高效进化
5.少样本蛋白质定向进化工具EVOLVEpro实操
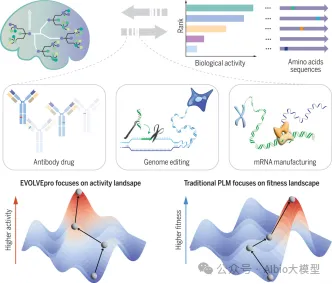
5.1. EVOLVEpro的背景与应用
5.2. 环境搭建与配置
5.3. 基于DMS数据的少样本微调
5.4. 基于实验数据反馈的少样本微调
5.5. 应用实例:Cas12f的高效进化
第五天基因编辑工具设计与挖掘案例复现
1. 设计MLH1 binder提高引导编辑编辑(PE)效率
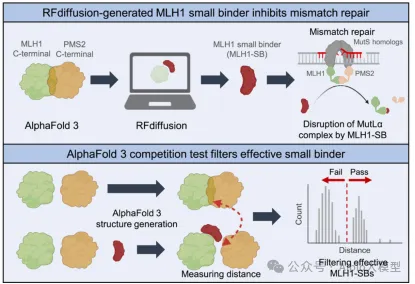
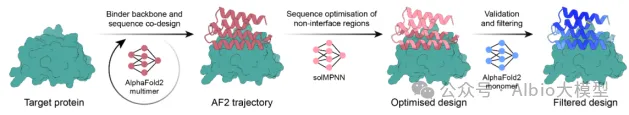
1.1. 背景知识:基于RFdiffusion + ProteinMPNN + AlphaFold的binder设计流程
1.2. 环境搭建与配置
1.3. 输入结构准备(AlphaFold预测)
1.4. 结构骨架生成:利用RFdiffusion进行结构采样与优化,生成蛋白质结构骨架
1.5. 序列设计:基于RFdiffusion生成的结构骨架,进行序列的优化设计
1.6.复合体结构预测验证:使用AlphaFold进行binder与目标蛋白复合体的结构预测,验证设计的复合体结构是否符合预期
1.7. 结果可视化:使用PyMOL进行结构和设计结果的可视化
2. Cas13抑制剂设计
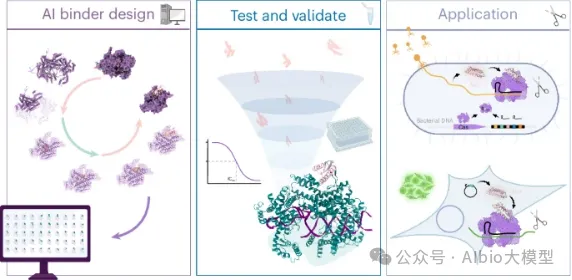
2.1. 背景知识:Cas13的结构与功能介绍
2.2. 输入结构准备
2.3. 蛋白质设计流程:结合RFdiffusion、ProteinMPNN与AlphaFold设计Cas13抑制剂
2.4. 设计结果分析和可视化
3.基于蛋白质语言模型挖掘新型CRISPR系统
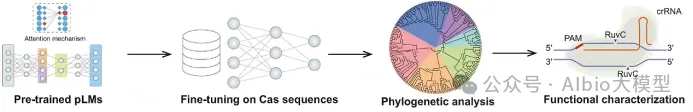
3.1. 蛋白质语言模型在酶挖掘中的介绍与流程
3.2. 序列数据库介绍与下载
3.3. 搜索(query)序列准备
3.4. 基于ESM语言模型挖掘Cas12家族基因编辑酶
4.基于三维结构挖掘新型CRISPR系统
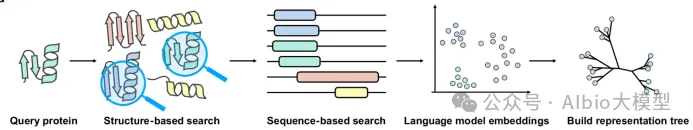
4.1. 结构比对的背景知识:结构比对的重要性与应用;比较不同结构比对工具的优缺点
4.2. Foldseek系列工具介绍:介绍Foldseek、Foldseek multimer、Folddisco、FoldMason等工具的基本原理和使用
4.3. 结构数据库介绍与下载:PDB,AFDB,ESM Atlas
4.4. 输入结构准备:准备用于比对的目标蛋白质结构文件
4.5. Foldseek网页版使用:演示如何使用Foldseek网页版进行结构比对;讲解如何理解输出结果并进行后续分析
4.6. Foldseek本地版使用:本地部署Foldseek并使用命令行工具进行比对
4.7. DALI和TM-align工具本地版使用:介绍DALI与TM-align工具本地版的安装与使用
4.8. 结构进化树构建:使用FoldMason构建蛋白质结构的进化树

主讲老师在学术界和工业界都有丰富算法开发和应用经验,博士毕业于国内顶尖课题组,从事蛋白质结构预测和蛋白质设计的研究工作,相关工作成果已在Cell Systems、Angew. Chem. Int. Ed.、JCIM等国际知名期刊发表论文。目前在知名药企担任高级研究员,主导AI驱动的大分子药物设计平台开发与团队管理。
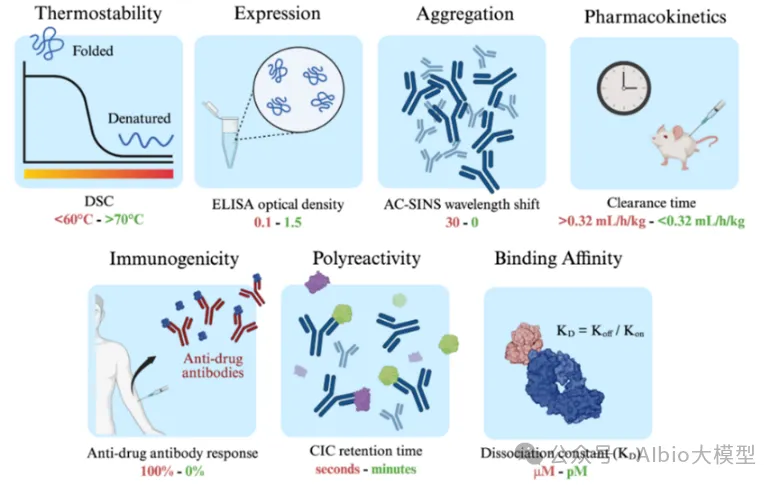
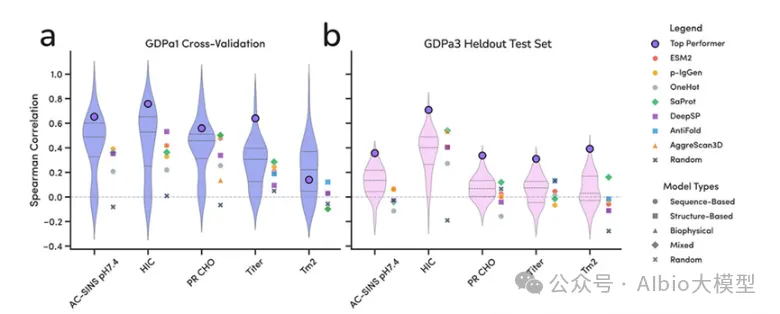
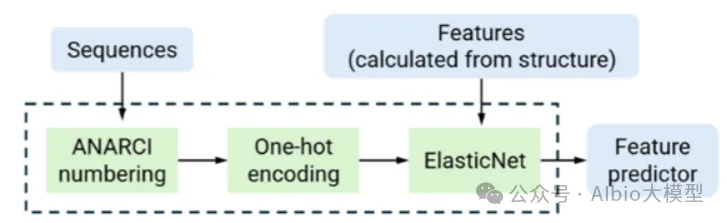

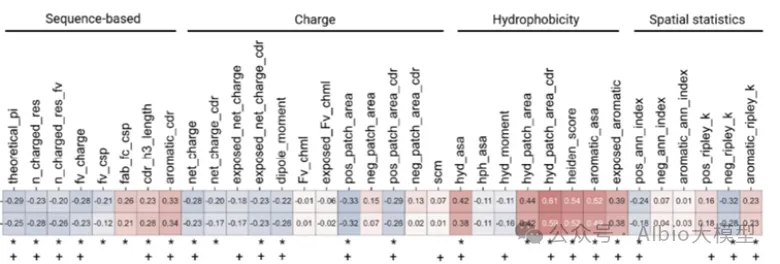
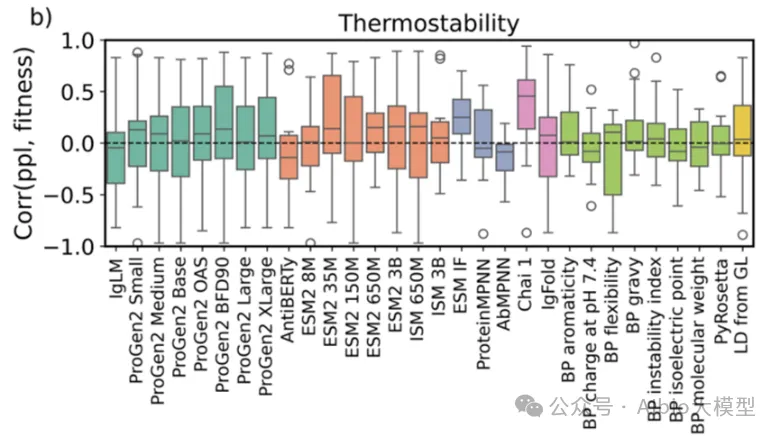
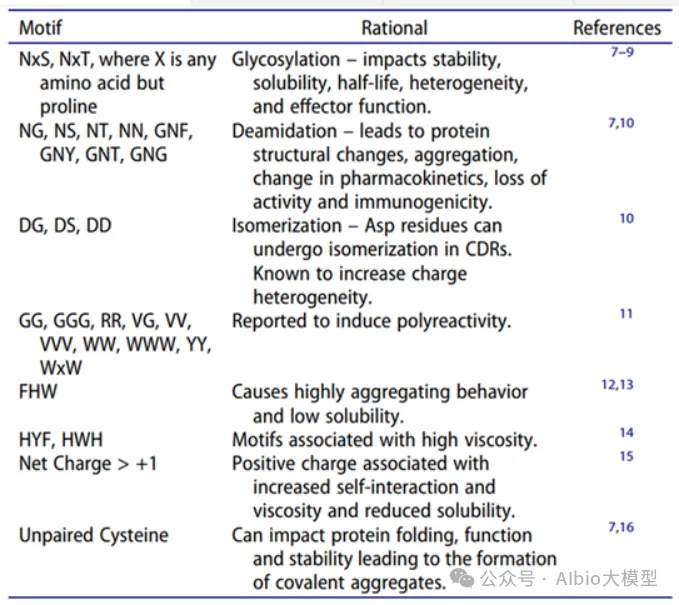


主讲老师在学术界和工业界都有丰富算法开发和应用经验,博士毕业于国内顶尖课题组,从事蛋白质结构预测和蛋白质设计的研究工作,相关工作成果已在Cell Systems、Angew. Chem. Int. Ed.、JCIM等国际知名期刊发表论文。目前在知名药企担任高级研究员,主导AI驱动的大分子药物设计平台开发与团队管理。
第一天:熟悉超算环境与蛋白质从头设计实践
1.环境搭建:Linux,VS code,Jupyter notebook*
a)超算的登录
b)Linux系统的常用shell命令:vim, ls, cd, less, rm等;
c)一些package安装的常用命令:pip, conda, source等。
d)Jupyter notebook的安装和使用。
e)VS code的基本配置:连接服务器;选择不同python版本的Interpreter;debug模式的使用等。
2.基础知识讲解
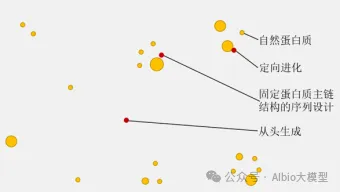
a)三类方法在不同程度上探索蛋白质序列空间:
i.蛋白质定向进化(directed evolution)
ii.固定蛋白质主链的序列设计(Fix-backbone protein design)
iii.蛋白质的从头设计(De novo protein design)
b)关键数据库:RCSB PDB, SCOPe, CATH, UniRef, BFD等
c)常见概念和名词: rotamer, scaffold, motif,domain,backbone,side-chain,apo和holo结构,
d)使用的不同模型的原理,transformer,diffusion模型,Flow Matching等。
3. Rfdiffusion3+ProteinMPNN生成序列
a)Rfdiffusion3生成蛋白质骨架结构,ProteinMPNN精细的生成氨基酸序列。
b)Rfdiffusion3的安装实操
c)Rfdiffusion3的使用实操
d)ProteinMPNN的安装实操
e)ProteinMPNN的使用实操
f)Rfdiffusion+ProteinMPNN生成序列,AphaFold2筛选序列。整体实操流程:
i.计算SAP(Spatial Aggregation Propensity)的值,选择3-6个氨基酸作为hotspot,即结合位点;这里需要使用Rosetta进行计算,首先将安装rosetta,准备蛋白,再计算每一个氨基酸的SAP值,将SAP数值映射到结构上。选择hotspot位点。
ii. Rfdiffusion结构设计,生成~10000个蛋白质主链结构;
根据上面挑选得到的hotspot位点,更改相应的hotspot参数,生成新的结构
iii.ProteinMPNN-FastRelax进行序列设计,每一个主链结构两个对应的序列,共设计~20000个序列;
iv.筛选:使用AlphaFold2预测设计结构,预测的置信度pAE<10,预测结构与设计结构的RMSD<1A,从中挑选95个进行实验验证。
4.其它的蛋白质设计方法的实操*
a)BindCraft——序列生成和筛选的自动化实现
BindCraft相比于Rfdiffusion+ProteinMPNN更加用户友好,一站式设计流程,序列的生成和筛选自动化实现。将讲解其中参数的设计和选择,如过滤序列条件、生成氨基酸的偏好性等。使用包括置信度评分(如AlphaFold2预测得到的pLDDT、ipTM)、物理指标(如Rosetta界面能量)和序列特征(如疏水性比例)进行筛选。
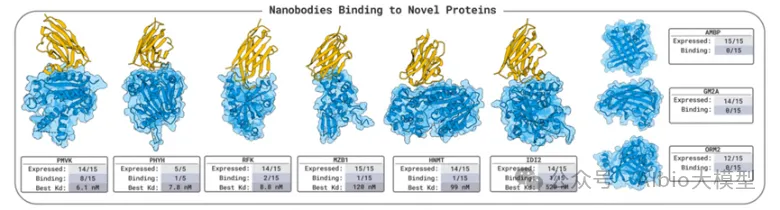
b)MIT开发的Bolzgen方法原理、安装使用讲解。
安装和使用boltzgen讲解,将详细讲解yaml配置文件的写法,以一个靶点为例,从头生成VHH与该靶点结合。
c)PPIFlow:基于flow-matching的生成方法,原理,安装和使用方法。
二、蛋白质结构预测和分析
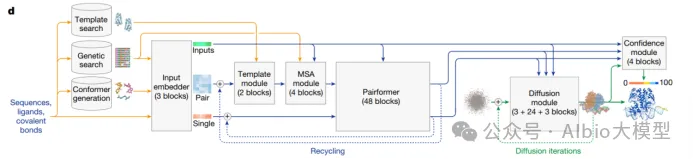
1.蛋白质结构预测方法
1)从CASP比赛结果来简述蛋白质结构预测方法的发展。基于能量函数 -> 接触图的应用 -> 端到端的预测结构(AlphaFold2)。
2)AlphaFold2的模型相比于以前的方法有什么改进
a)将基于MSA和基于模板的方法整合,使用注意力机制进行MSA信息和模板信息的相互交流。
b)以前提取MSA信息为计算协方差矩阵 ,AlphaFold2创造性的直接将MSA信息作为输入,将图像识别的算法转变成了自然语言处理算法,减少了中间处理过程中的信息损失。
3)AlphaFold3相比于AlphaFold2改进了什么,还有什么不足。
a)扩展到了多种生物分子的复合物结构预测,包括蛋白质-DNA、蛋白质-RNA、蛋白质-小分子,并使用扩散模型。
b)复合物组装与动态预测缺陷,抗体-抗原复合物结构准确度有待提高。
4)运行网页server上的AlphaFold3预测结构
5)如何使用AlphaFold3预测蛋白质的糖基化,不同糖基化的类型的输入方法。
6)AlphaFold3输出结果分析,各项置信度指标的含义,以及如何判断预测的准确度,如pLDDT,ipTM,PTM,PAE。
7)本地部署和运行ColabFold,由于AlphaFold3在安装过程中需要下载大量资源,且不能商用,因此不演示AlphaFold3的安装过程,如有问题可以帮助解决。
2.蛋白质结构分析和可视化
1)pdb文件的解读,每一行中的内容代表什么含义。
2)用 pymol 可视化蛋白质结构*
a)pymol的基础操作讲解
b)如何将实验值投影到结构图的颜色上,如何画出发表文章中好看的图
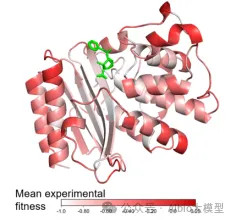
3)计算蛋白质结构中两个氨基酸的距离*
a)使用python的文本文件操作实现
b)使用python中biopython包实现
3.蛋白质结构相关物理性质的计算*
1)二级结构的分类和计算
2)溶剂可及表面积(SASA)的讲解及计算
三:蛋白质序列分析,数据挖掘和训练数据准备
讲解和实操:
1.获得同源序列
1)了解不同蛋白质序列库,如UniRef90,UniClust30,Pfam等
2)了解不同工具原理并使用:NCBI BLAST,Jackhmmer,HHblits
3)给定一条蛋白质序列,比对序列库,生成多序列比对(MSA)*
从AlphaFold2的经典代码仓库中找到它的生成MSA的代码并学习(alphafold/alphafold/data/tools/jackhmmer.py)。
运行示例:jackhmmer –cpu 8 -N 2 -E 1e-7 query.fasta uniprot_sprot.fasta -o output.sto
2.对MSA进行频率分析*
1)使用python的文本文件操作实现
2)使用python中biopython包实现
3)绘制序列Logo,可视化的展示每个位点的氨基酸频率和保守性
3.序列的同源性计算和进化树的绘制*
1)不同同源性的计算方法及应用情景,氨基酸序列的identity和Similarity,BLOSUM62的介绍。
2)进化树的绘制
4.基于序列相似性阈值划分训练集和测试集*
1)为什么要做?避免数据泄露
2)选择相似性度量方法
3)相似性矩阵的计算
4)划分数据集
5.大规模蛋白质序列的聚类分析和去冗余*
1)为什么要做?防止过度学习某一类序列特征,消除序列偏差;也能防止训练过程中数据泄露。
2)聚类方法的选择,CD-HIT、MMseq2和Linclust
3)选择代表序列,去冗余
4)实际复现S2ALM这一模型文章中的聚类方法。mmseqs easy-cluster examples/DB.fasta clusterRes tmp –min-seq-id 0.7 -c 0.8 –cov-mode 1
四、蛋白质的大语言模型及其应用
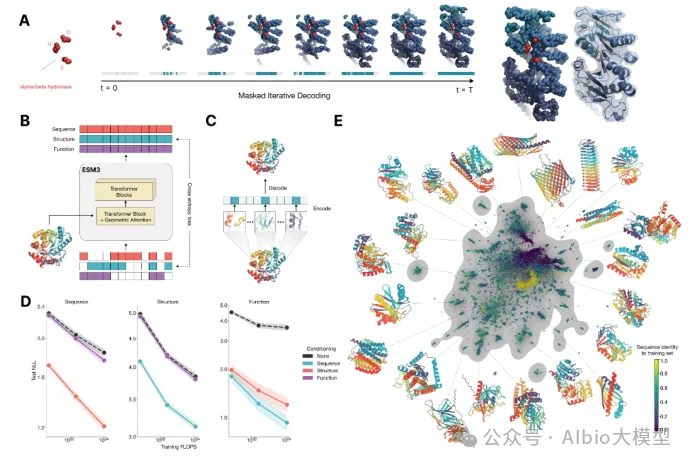
1.基础知识讲解
1)介绍蛋白质的语言模型(26字母语言模型->20氨基酸字母表,上下文依赖->氨基酸的共进化)
2)为什么要开发蛋白质大语言模型?1. 相比于结构或功能信息,序列信息更加海量;2. 蛋白质序列通过进化而来,可以学习蛋白质基本规律,折叠,共进化等
3)模型架构和基础理论:transformer,多头注意力机制,Bert,GPT,T5等
2.基于Bert架构的蛋白质语言模型
1) ESM系列(ESM-1b、ESM-1v、ESM2、ESM C)
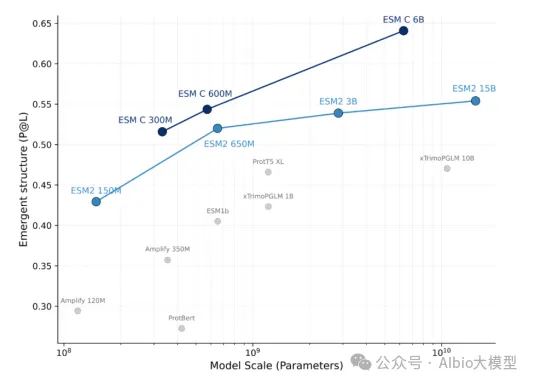
2)ESMFold:无需MSA信息的结构预测
3)使用抗体序列库训练的语言模型:Ablang,AntiBERTy
3.类似GPT的生成模型ProGen
1)36层Transformer解码器架构,包含12亿参数
2)引入“控制标签”(如蛋白质家族ID、功能属性)作为输入,生成蛋白质序列空间以外的新的蛋白质序列
3)成功生成新的溶菌酶
4.多模态的蛋白质语言模型ESM3
1)模型架构融合序列,结构和功能信息
2)相比于ESMFold,单体结构预测精度更好
3)基于多模态提示(序列、结构、功能关键词)设计新的蛋白质序列
4)ESM3的安装,生成序列,快速结构预测。*
5.蛋白质语言模型的应用和实战演练*
1)获得序列embedding以构建下游模型(Cell systmes文章举例),从文章github仓库中提炼序列embedding的代码并学习使用。
看懂代码中EncodingGenerator的类,将这个类方法用在我们自己的代码上,实现蛋白质序列的不同方式encoding,包括”onehot”, “georgiev”, “esm”系列模型。
2)使用不同的蛋白质语言模型,零样本的预测蛋白质突变效应。
3)给定少量的突变效应数据作为训练数据,训练模型,预测新的突变效应值。
五、深度学习辅助酶设计
1.基础知识讲解
酶的过渡态理论,theozyme,fitness landscape,epistasis
2.酶学性质预测
1.DLKcat与GotEnzyme数据库介绍
2.UniKP:利用预训练模型挖掘、改造Kcat
3.CLEAN:基于对比学习的EC号预测挖掘稀有脱卤酶
3.蛋白质热稳定性改造
1.MutCompute介绍
2.利用MutCompute改造PETase(Nature)
3.ThermoMPNN介绍与使用*
4. Pythia介绍与使用*
4.从Frances H. Arnold(2018年因在酶的定向进化领域的贡献获得诺贝尔化学奖)的工作看酶的定向进化方法的发展
1.传统定向进化实验流程
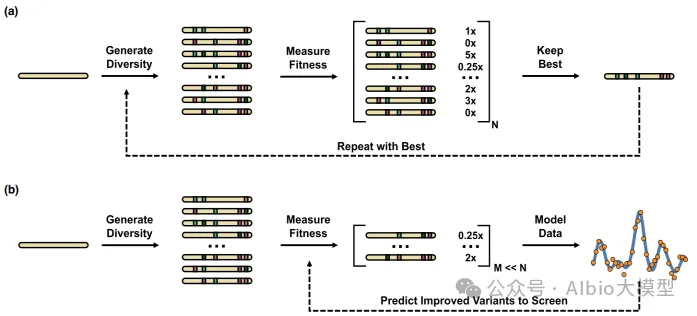
2.MLDE(Mechine Learning Directed Evolution), 学习序列与酶性能之间的映射关系,推荐新的突变组合(PNAS文章)
3.ftMLDE(focused training MLDE),主动学习流程,构建informative的训练数据(Cell Systems文章)。零样本突变效应预测挑选数据集,再通过小样本数据训练的策略微调。
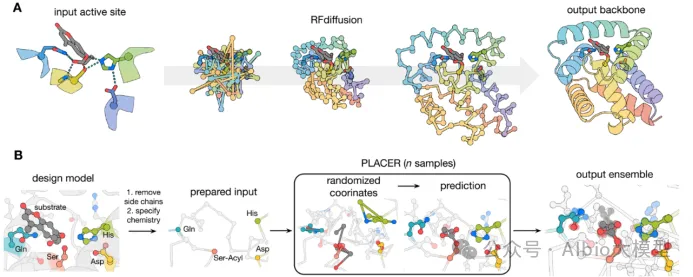
5.酶的从头设计
1.从头设计Diels-Alder催化酶
a)基于Rosetta的Inside-out策略(Science文章)
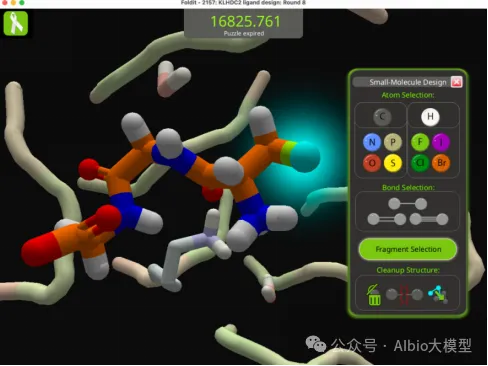
b)通过Foldit蛋白质折叠游戏改善结构问题(Nat. Biotechnol.文章);
c)Foldit蛋白质折叠游戏的实践*
2.从头设计荧光素酶,Family-wide hallucination,基于该酶家族的结构幻化出新的结构(Nature文章)
3.RFdiffusion+PLACER从头设计丝氨酸水解酶(Science文章)
6. 利用预测结构的相似性,挖掘序列的新酶功能(复现顶刊cell文章)*
1.InterPro数据库中下载数据
2.TM-score计算结构距离
3.UPGMA结构聚类,画出进化树
4.挑选序列
六、蛋白质功能与互作预测;实验验证与AI模型训练预测闭环
1.蛋白质功能预测:
1)基础知识:
a)基因本体论(Gene Ontology, GO),
b)MF/BP/CC,MF Molecular Function分子功能;BP Biological Process生物过程;CCCellular Component 细胞组分。
c)GAF (GO Annotation File) 文件。
d)本体文件来理解GO术语之间的层次关系。
e)解析GAF,提取蛋白质ID和GO ID。
2)DeepGO-SE,通过蛋白质的语言模型提取序列嵌入,预测蛋白质的功能
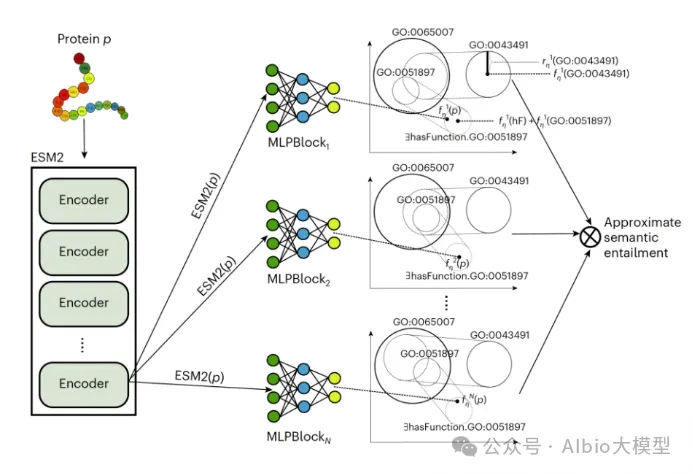
3)DPFunc:先用蛋白语言模型提取残基特征,再在接触图上用 GCN 学习结构信息,并引入结构域(domain)指导,最后把多层特征映射到 GO 图上,显著提升对罕见功能项和低序列相似蛋白的预测精度
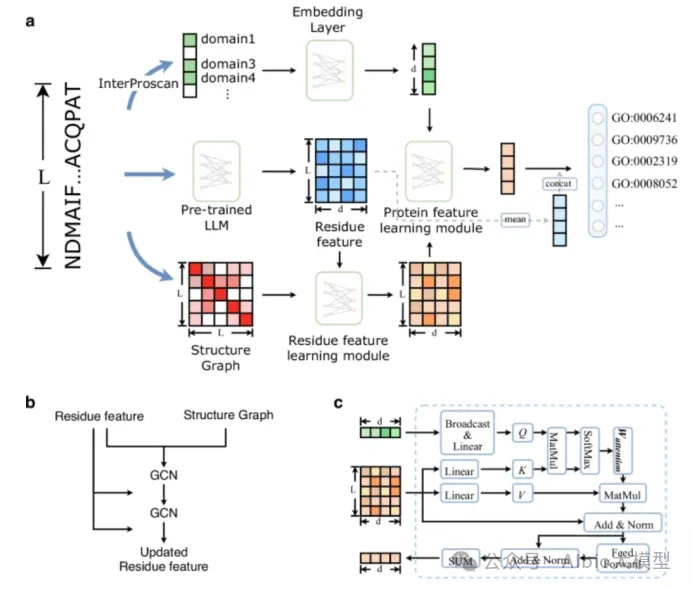
4)Prot2Text-V2模型。Prot2Text-V2将图神经网络(Graph Neural Network, GNN)与大型语言模型(Large Language Model, LLM)融合到同一个编码器-解码器框架中,有效整合了包括蛋白质序列、结构和文本注释在内的多种数据,以自由文本形式输出蛋白质功能预测结果
5)ProteinKG65构建蛋白质知识图谱,基于Gene Ontology (GO) 和 UniProt 等权威知识库,将蛋白质的功能、结构、相互作用等知识组织成图谱形式,支持下游的机器学习任务,如蛋白质功能预测、表示学习、药物靶点发现等
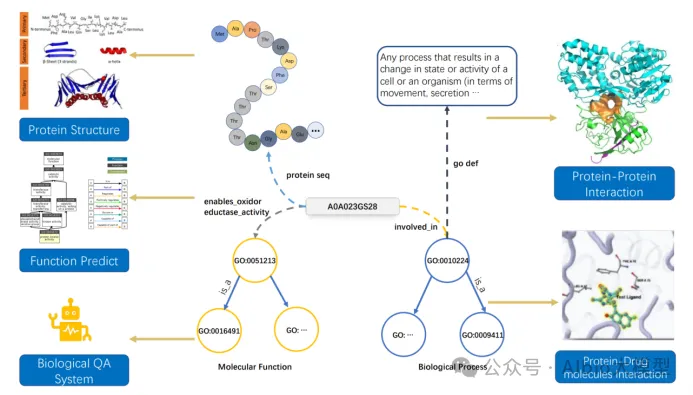
2.蛋白质相互作用预测:
Science文章:使用更深的进化信号:omicMSA+新的深度学习网络:RF2‑PPI。在全人类蛋白质组中筛出一批高置信度的互作,用于补齐人类互作图谱、解释疾病突变和蛋白功能。
1. 更深的进化信号:omicMSA
从约 30 PB 的未组装基因组/转录组数据里挖人类蛋白的同源序列,而不仅仅依赖 UniRef 等传统数据库。
构建omicMSA,使得每个蛋白的深度比常规模板 MSA 深 7 倍左右,协同进化信号显著增强。
2. 新的深度学习网络:RF2‑PPI
基于 RoseTTAFold2 框架开发了一个新的 PPI 预测网络 RF2‑PPI,用来快速估计两条蛋白是否互作以及界面大致形态。
为了训练 RF2‑PPI,构建了很大的数据集:从约 2 亿个预测蛋白结构中抽取各种结构域组合,构建了大规模的 DDI 训练样本,使训练集规模相比传统 PPI 结构数据扩大约 16 倍
筛选流程:
1. 人类蛋白集合
取约 19,500 个人类蛋白序列(UniProt 等),所有可能的配对约 2 亿对。文章中实际筛查约 2 亿对蛋白组合。
2. 构建深度 omicMSA
对每个蛋白,以及蛋白对,基于 30 PB 基因组/转录组数据构建 omicMSA,并对每个蛋白对生成配对 MSA(pMSA),用于协同进化分析和后续深度学习输入。
3. 快速预筛:共进化 / RF2‑PPI 粗打分
先用直接耦合分析(DCA)等共进化方法,结合 RF2‑PPI 对 2 亿对蛋白打一个“互作概率”分数(RFIntProb),过滤掉大部分不可能的组合。
从 4360 万对预筛后的蛋白对中,用 RF2‑PPI 进一步筛选出约 190 万对 RFIntProb > 0.3 的候选。
4. 精细建模:AlphaFold2 复合物结构
对这约 190 万对蛋白,用 AlphaFold2(多聚体/复合物模式)进行结构预测,得到每一对的三维复合物模型以及一个基于界面质量的互作概率(AFIntProb)。
根据 AFIntProb 以及界面大小等指标选择高置信度互作。
5. 高置信度集的定义
在所有蛋白对中,最终在“完全无先验”的全 2 亿对筛选中得到 6,763 个高置信度 PPI;
进一步结合已有数据库(STRING、BioGRID、UniProt 里有物理互作证据的 115 万对蛋白对),在有先验证据的集合上又识别出 21,960 个高置信度 PPI。
综合各种来源和精度阈值,共预测出 17,849 个 PPI,预期精度约90%,其中 3,631 个此前实验未报道的新互作。
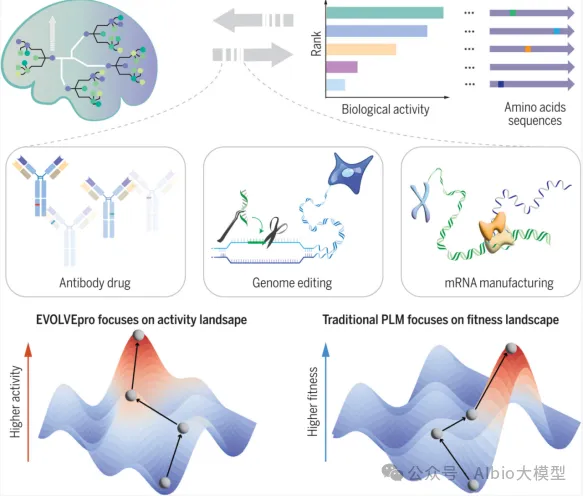
3. AI模型训练预测和实验闭环
以 EVOLVEpro 为例,实践计算–实验闭环:
1.初始化
●选取少量已测序列(野生型 + 文献或少量自设计突变),测定活性。
●用蛋白语言模型把序列编码成向量,训练一个初始的监督回归模型(序列向量→ 活性)。
2.生成候选序列
●设定允许的突变范围(允许 1–3 点突变、限定在特定位点/区域)。
●在该空间内大规模生成候选序列(10^3–10^5),可结合 embedding 空间附近搜索、局部扰动等策略。
3.预测与智能选样
●用回归模型对所有候选序列预测活性或综合评分。
●依据主动学习策略挑出一小批要做实验的序列:
●直接选预测值最高的 top‑k;或
●结合预测不确定性、序列多样性等,使样本既“高潜力”又“信息量大”。
4.实验验证
●合成/构建这批候选序列,利用高通量实验(如流式、板读、NGS 条形码筛选等)测定真实活性。
●得到新一轮“序列–活性”数据。
5.回流更新与迭代
●将新数据并入训练集,重新训练或微调回归模型(PLM 一般保持不变)。
●重复“生成候选 → 预测选样 → 实验验证 → 更新模型”的循环,通常 3–4 轮即可显著提升目标性能。

主讲老师博士毕业于某合成生物学专业顶尖双一流高校,主要从事合成生物学工具开发,基因电路设计与动态调控,高附加值天然产物化学品合成路径挖掘与高水平合成,精通大肠杆菌,酿酒酵母,毕赤酵母,解脂酵母等微生物细胞工厂的基因编辑和构建,具备完整的从上游菌株改造到下游放大生产的产业化经验,已经实现多个产品的产业化落地,在Metabolic Engineering,Bioresour Technol,Appl Microbiol Biotechnol,J Agric Food Chem,ACS Synthetic Biology等杂志共发表SCI文章16篇,申请发明专利8项
一、:合成生物学导论与入门
主题:从DNA组装到生命系统设计
一、合成生物学定义与发展简史(1小时)
定义与核心概念
合成生物学是通过工程化方法设计和构建生物系统,以解决实际问题的跨学科领域,融合生物学、工程学和信息学。
核心目标:改写生命遗传指令,实现定制化功能(如生产药物、能源)。
发展简史
起源:20世纪中叶,DNA双螺旋结构发现和蛋白质合成技术奠定基础。
里程碑:
2000年:基因网络开关设计(Collins团队)。
2002年:人工合成脊髓灰质炎病毒(Wimmer团队)。
2010年:首个人工合成基因组细胞(Venter团队)。
2014年:非天然碱基配对整合(Romesburg团队)。
现状:21世纪后快速发展,聚焦基因组设计、细胞工程和产业应用。
二、常用软件工具与网站介绍
基因设计工具
DNAWorks:免费在线软件,用于设计寡核苷酸链(适用小片段合成)。
商业软件:如Snapgene,GenBank(序列数据库)、EMBL(欧洲生物信息学资源),支持基因组全序列下载和分析。
功能:序列优化、引物设计、模拟基因表达。
代谢通路建模工具
KEGG(京都基因与基因组百科全书):可视化代谢通路,辅助设计合成生物学模块。
实践平台
iGEM(国际基因工程机器大赛)官网:提供标准化生物元件库和社区资源。
NCBI(美国国家生物技术信息中心):综合数据库,支持基因序列检索和功能注释。
三、代谢数据库与知识库
核心数据库
代谢组学数据库:如HMDB(人类代谢组数据库),整合代谢物结构和功能信息。
基因组数据库:GenBank、EMBL、DDBJ(日本DNA数据库),存储全基因组序列。
功能:通过序列比对和通路映射,预测基因功能和代谢网络。
知识库应用
设计阶段:利用数据库筛选标准化生物元件(如启动子、终止子),确保设计可行性。
测试阶段:比对实验数据与数据库,验证代谢通路效率(如酶活性分析)。
四、互动实践:常用软件使用
实践目标
掌握DNA序列设计、组装模拟。
步骤与工具
DNA设计:使用Snapgene输入目标序列,生成寡核苷酸链并模拟组装。
数据分析:通过NCBI BLAST比对序列相似性,评估设计准确性。
二、基因编辑与工具技术
eCRISPR技术、基因合成、生物元件设计(启动子/终止子)
一、基因编辑技术基础概念
基因编辑定义与核心原理
定义:通过人工干预修改生物体基因组,实现特定性状改变。
核心原理:
DNA断裂与修复:双链断裂(DSB)触发细胞修复机制(NHEJ或HDR)。
碱基编辑:直接修改单个碱基,无需断裂DNA。
基因编辑工具发展历程
第一代:ZFN(锌指核酸酶,2000年代初,靶向性差)。
第二代:TALEN(转录激活因子样效应核酸酶,2010年代,灵活性提升)。
第三代:CRISPR-Cas9(2012年诺贝尔奖,高效、低成本、可编程)。
二、CRISPR-Cas9系统详解
CRISPR系统组成与工作机制
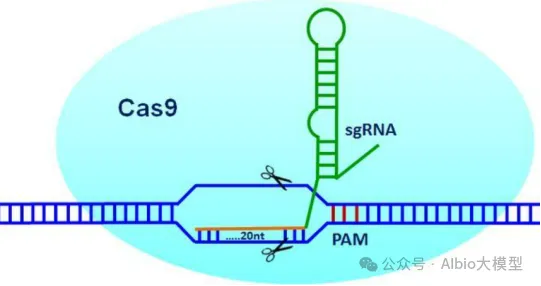
核心组件:
Cas9蛋白:切割DNA的“剪刀”。
sgRNA(单导RNA):引导Cas9到目标位点(含20nt互补序列)。
PAM(原间隔序列):Cas9识别的短序列(如NGG)。
工作机制:
sgRNA与Cas9结合,形成复合物。
复合物识别PAM,切割DNA双链。
细胞通过NHEJ或HDR修复断裂。
CRISPR系统操作流程
步骤:
设计sgRNA:选择目标基因的PAM序列,设计20nt互补RNA。
构建载体:将sgRNA和Cas9基因插入质粒(如pCRISPR1)。
转化宿主:将载体导入细胞(如HEK293T细胞)。
筛选与验证:通过PCR、测序确认编辑效率。
CRISPR技术优化方向
提高特异性:使用高保真Cas9变体(如HF-Cas9)。
降低脱靶率:优化sgRNA浓度,避免非特异性切割。
扩展应用场景:开发CRISPR-Cas12(靶向单链DNA)和CRISPR-Cas13(靶向RNA)。
CRISPR实验注意事项
实验设计:设置阴性对照(如非靶向sgRNA)。
数据分析:使用NGS(下一代测序)评估编辑效率。
三、基因编辑实验设计实践
实验方案设计要点
明确目标:编辑单个基因(如敲除)或多基因(如代谢通路优化)。
选择宿主:根据基因功能选择模式生物(如大肠杆菌、酵母、人类细胞)。
优化条件:调整sgRNA浓度、Cas9表达量、转化方法(如电穿孔)。
不同微生物宿主SgRNA设计原则
原核生物(如大肠杆菌):
优先选择PAM序列(如NGG),避免CRISPR-Cas系统的天然防御机制。
真核生物:
避免设计在基因组重复区域或调控序列中的sgRNA。
筛选方法与验证
筛选:通过抗生素抗性或荧光标记(如GFP)筛选成功转化细胞。
验证:PCR扩增:设计引物跨越编辑位点,检测片段大小。
测序:对PCR产物进行Sanger测序,比对参考序列。
功能检测:如编辑后基因表达量(qPCR)、表型变化(如细胞生长速度)。
单基因编辑设计与多基因编辑设计
单基因编辑:
步骤:设计sgRNA→构建载体→转化细胞→筛选→验证。
多基因编辑:
示例:在酵母中同时编辑3个代谢基因(如ADH1、PGK1、GAPDH)。
第三天
三、基因线路工程与动态调控
主题:细胞内的“逻辑电路
基因电路设计原理
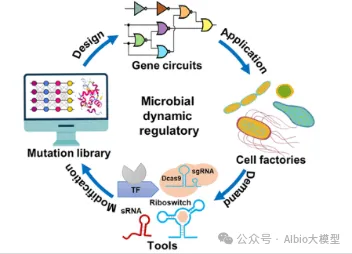
一、基因线路概述
1. 定义与功能
o基因线路:生物体内基因表达的调控网络,通过逻辑门(与门、或门、非门)实现特定功能(如代谢调控、信号响应)。
o核心功能:
§开关控制:基因表达的“开/关”(如乳糖操纵子)。
§信号处理:环境信号(如光、温度)的响应与转导。
§稳态维持:通过负反馈调节基因表达水平。
2. 应用领域
o生物制造:优化代谢通路。
o疾病治疗:基因疗法。
o环境监测:工程菌检测污染物。
3. 案例对比
o原核案例:大肠杆菌乳糖操纵子(LacI蛋白抑制转录,乳糖诱导表达)。
o真核案例:人类β-珠蛋白基因增强子(远端调控序列激活转录)。
二、基因线路设计原则
1. 模块化设计
o原则:将复杂功能拆解为独立模块(如启动子、转录因子、报告基因)。
o示例:设计“光控开关”线路,分离光敏蛋白与报告基因(如GFP)。
2. 稳定性与可预测性
o正交设计:减少模块间干扰(如避免共用转录因子)。
o鲁棒性:通过冗余设计(如双启动子)确保功能稳定。
3. 实验验证方法
o荧光报告基因:定量表达水平(如GFP荧光强度)。
oqPCR:检测转录效率(如mRNA量)。
三、实践操作:基因线路构建
1. 工具介绍
oCRISPR-Cas9:精准编辑基因(如敲除抑制子)。
o质粒载体:携带基因线路元件(如pCRISPRi)。
o电转化技术:将载体导入细胞(如大肠杆菌)。
2. 设计“光控开关”基因线路
o步骤:
1. 设计光敏蛋白:选择光敏离子通道(如ChR2)或光敏转录因子(如PhyB)。
2. 构建载体:将光敏蛋白基因与报告基因(如GFP)插入质粒。
3. 转化宿主:将载体导入大肠杆菌,筛选阳性克隆。
4. 验证功能:光照后检测GFP荧光(定性)或qPCR(定量)。
3. 实验
o阴性对照:使用非光敏蛋白(如GFP空载质粒)。
o优化条件:调整光强、曝光时间。
四、动态调控原理
1. 负反馈与正反馈
o负反馈:转录因子抑制自身表达(如乳糖操纵子中的LacI蛋白)。
o正反馈:转录因子激活自身表达(如噬菌体λ的CI蛋白)。
2. 时间延迟效应
o原因:基因表达与调控的滞后(如转录、翻译过程)。
o影响:导致系统振荡或稳态偏离。
3. 案例:大肠杆菌动态调控高产莽草酸
o背景:莽草酸是合成抗病毒药物的原料。
o调控机制:
§负反馈:莽草酸合成酶(如AroB)抑制自身表达。
§优化策略:通过CRISPR敲除抑制子(如AroB的负调控蛋白),提高产量。
五、系统集成与案例分析(
复杂线路设计策略
o振荡器:结合负反馈与时间延迟(如基因表达振荡)。
o开关:利用逻辑门(如与门)控制多基因表达。
o脉冲发生器:通过瞬时信号触发基因表达(如热激响应)。
1. 案例分析:合成生物学中的动态调控
第四天
四、代谢工程与生物制造
主题:微生物细胞工厂的理性设计与代谢通路设计与重构
一、细胞工厂与理性设计范式
1. 细胞工厂定义
o利用工程化微生物(如大肠杆菌、枯草芽孢杆菌、酵母)作为“生物反应器”,通过重构代谢网络生产高值化学品(如1,3-丙二醇、氨基酸、生物燃料)。
2. 范式转型
o传统模式:随机诱变+高通量筛选(低效、不可预测)。
o理性设计:基于基因组尺度模型 + 代谢通量分析 + AI预测(精准、可复现)。
3. 发展历程
o天然发酵(酿酒酵母产乙醇)→ 代谢工程(大肠杆菌产乳酸)→ AI驱动设计(AlphaFold辅助酶结构预测,优化限速步骤)。
4. 核心挑战
o鲁棒性:抗渗透压、高温、产物毒性(如1,3-丙二醇抑制生长)。
o效率:产物得率,需突破热力学极限。
o原料多样性:利用农业废弃物(如秸秆水解液)替代葡萄糖,降低碳源成本。
二、物质流-能量流-信息流协同设计
1. 热力学驱动:ATP/NADH平衡
o产物合成需消耗还原力(如NADPH用于脂肪酸合成)或产生还原力(如1,3-丙二醇生成消耗NADH)。
o策略:引入NADH再生系统(如甲酸脱氢酶)或切换碳源(甘油 vs 葡萄糖)调控辅因子比例。
2. 动力学驱动:酶活性调控
o限速酶(如AroE、DhaT)表达量不足导致通量瓶颈。
o优化方法:使用NCS文库(N端编码序列)精细调控翻译效率,提升酶活性3–8倍。
3. 代谢网络重构:通量平衡分析(FBA)
o原理:基于质量守恒与反应约束,求解最大生物量或产物产量的代谢流分布。
4. 案例:碳-氮比调控谷氨酸棒杆菌产谷氨酸
o高碳氮比(>20:1)激活谷氨酸脱氢酶,抑制TCA循环,使α-酮戊二酸积累并转化为谷氨酸。
三、底盘细胞开发策略
1. 设计原则
o鲁棒性底盘:引入热休克蛋白(如GroEL/ES)增强耐热性,提升高温发酵稳定性。
o稳定性底盘:基因组简化(删除非必需基因如 prophage、转座子),减少代谢负担与基因组不稳定性。
2. 技术方法
o智能抗逆元件:构建温度响应型启动子,在37°C以上激活抗逆基因表达。
o无诱导表达系统:利用组成型强启动子替代IPTG诱导,降低生产成本。
3. 案例:枯草芽孢杆菌底盘改造
o目标产物:N-乙酰神经氨酸(Neu5Ac)
o改造策略:
§引入唾液酸合成途径(neuA, neuB, neuC)
§构建NCS文库优化关键酶表达(GFP荧光强度提升8.47倍)
§删除竞争途径(如glcA)减少副产物
第五天
五、 合成生物学中高通量筛选技术
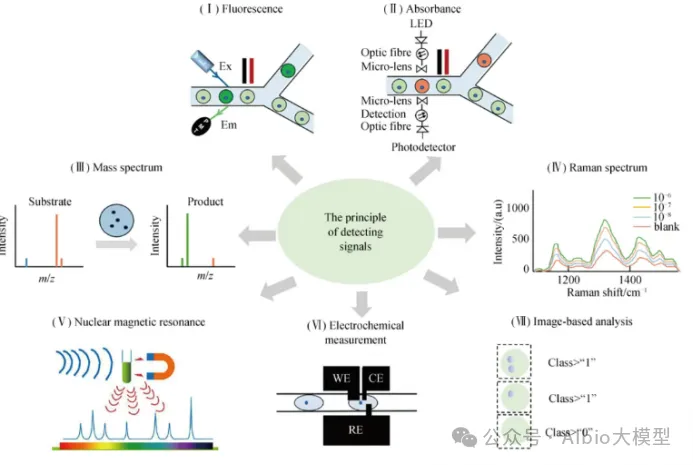
1、主题:传统高通量筛选技术
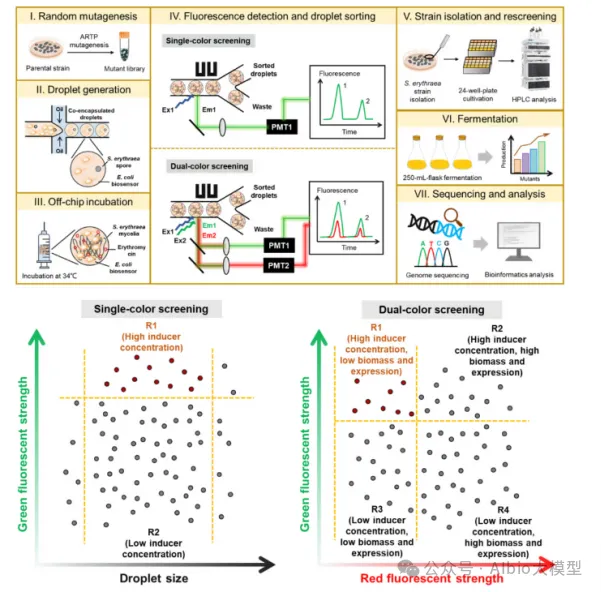
一、传统高通量筛选技术体系
1. 三大技术支柱
o机器人自动化系统:通过协作机器人(如Explorer G3)实现96/384孔板的自动加样、温孵与转移,日处理通量可达10⁵–10⁶样品。
o液体处理器:精准控制纳升–微升级液体分配(误差<2%),支持混合、稀释、分液一体化,消除人为操作偏差。
o检测系统:
§荧光检测:报告基因(GFP、LacZ)用于基因表达水平量化;
§细胞增殖检测:MTT/Resazurin法评估细胞代谢活性;
§离子通道筛选:膜片钳自动化平台检测神经靶点化合物活性。
2. 数据处理流程
o原始数据:荧光强度、吸光度、成像特征
o标准化:Z’因子评估(Z’>0.5为合格)
o分析工具:GraphPad Prism、Python(pandas + scikit-learn)进行剂量响应曲线拟合与Hit筛选。
3. 案例
o报告基因筛选:构建“GFP-乳糖操纵子”大肠杆菌库,用荧光酶标仪筛选强启动子变体。
二、微流控与液滴微流控技术
1. 技术原理
o微流控芯片:通过光刻/软光刻技术在PDMS芯片中构建微通道网络,集成样品制备、反应、分选、检测单元(尺寸<2 cm²)。
o液滴微流控:利用油水两相流生成皮升级(pL)单分散液滴,作为独立微反应器,实现:
§单细胞包裹与恒化培养
§酶基因表达产物的高通量筛选
§细胞裂解与代谢物捕获
2. 通量优势
o传统:10³–10⁴样品/天
o液滴系统:10⁵–10⁶液滴/小时(DropAI系统实测)
3. 实验设计
o非标记荧光分选:利用微生物自发荧光(NADH/FAD)检测生长速率,分选“高产”菌株。
o荧光编码系统:FluoreCode技术,通过不同荧光强度组合编码液滴组分,实现百万级组合并行筛选。
三、拉曼光谱在代谢物高通量筛选中的应用
1. 原理与优势
o拉曼散射:激光激发分子振动模式,产生特征“指纹光谱”,无需标记即可检测:
§脂肪酸(C-H伸缩峰:2850 cm⁻¹)
§聚羟基脂肪酸酯(PHAs,1240 cm⁻¹)
§蛋白质二级结构(Amide I, 1650 cm⁻¹)
o无损、快速、单细胞级:单细胞光谱采集<1秒,适用于活细胞动态监测。
2. 操作流程
o样品准备:细胞悬液滴于硅基片或微流控出口
o光谱采集:使用532 nm或785 nm激光,积分时间1–10 s
o数据分析:
§主成分分析(PCA)区分细胞表型
§支持向量机(SVM)分类高产/低产菌株
3. 应用
o油脂生产菌筛选:对产油酵母(如Yarrowia lipolytica)进行拉曼成像,识别高脂含量单细胞。
o液滴-拉曼联用:SERS增强基底嵌入微流控芯片,实现“生成-检测-分选”一体化。
4. 技术瓶颈
o信号弱(需SERS增强)
o数据维度高(>1000波数点/光谱),需AI降维分析
四、AI驱动的高通量筛选闭环
1. DBTL循环升级
oDesign:AI预测酶结构(AlphaFold)→ 优化催化位点
oBuild:自动化合成基因库(CRISPR-Cas9 + Golden Gate)
oTest:液滴微流控 + 拉曼/荧光检测 → 生成百万级表型数据
oLearn:机器学习模型(XGBoost、神经网络)训练预测模型,反向优化设计
2. 工业级平台案例
oSynGears™平台:AI驱动的“数字基座”,整合基因设计、通路模拟与筛选数据,实现“设计即优化”。

主讲老师刘老师,生物信息学博士,从事医学生物信息及人工智能研究 15年,曾在新 加坡基因组研究院及美国加州大学洛杉矶分校研究多组学数据在复杂疾病诊疗中的应 用。研究领域涉及人工智能、自然语言处理、功能基因组学、宏基因组学、转录组学、miRNA及靶基因网络分析,单细胞测序数据分析,基因调控网络时序分析,蛋白质互 作网络分析,多组学联合分析等。主持省级自然科学基金等项目 4项,开发过数个生 物信息学工具,发表 SCI论文 20余篇,其中人工智能算法文章 10余篇,编著医学数 据分析实用教材一部。
第三天、深度学习识别疾病标志物上午深度学习模型识别疾病标志物 Python代码解析及 GPU服务器上机实操

主讲老师来自985高校神经科学博士,主要利用代谢组学、转录组学和分子生物学等技术研究神经内科慢性病的发病机制和生物标志物。擅长高效液相色谱-质谱联用(LC-MS)技术进行非靶向和靶向代谢组学从样本制备到数据分析的全流程研究,以及多组学大数据的生物信息学整合分析。5年内在J Clin Invest, EBioMedicine, Cell Death Dis, Cell Death Discov, Nanotoxicology等杂志发表SCI论文!
第一天上午
A1 代谢物及代谢组学的发展与应用
(1) 代谢与生理过程;
(2) 代谢与疾病;
(3) 非靶向与靶向代谢组学;
(4) 空间代谢组学与质谱成像(MSI);
(5) 代谢组学与药物和生物标志物;
(6) 代谢流与机制研究。
A2 代谢通路及代谢数据库
(1) 几种经典代谢通路简介;
(2) 三大常见代谢物库:HMDB 、METLIN 和 KEGG;
(3) 代谢组学原始数据库:Metabolomics Workbench 和 Metabolights. A3 参考资料推荐
第一天下午
A4 代谢组学实验流程简介
A5 色谱 、质谱硬件与原理解析
(1) 色谱分析原理与构造;
(2) 色谱仪和色谱柱的选择;
(3) 色谱的流动相:梯度洗脱法;
(4) 离子源、质量分析器与质量检测器解析;
(5) 质谱分析原理及动画演示;
(6) 色谱质谱联用技术(LC-MS);
第二天上午
B1 代谢物样本处理与抽提
(1) 各种组织、血液和体液等样本的提取流程与注意事项;
(2) 代谢物抽提流程与注意事项;
(3) 样本及代谢物的运输与保存问题;
B2 LC-MS 数据质控与搜库
(1) LC-MS 实验过程中 QC 和 Blank 样本的设置方法;
(2) LC-MS 上机过程的数据质控监测和分析;
(3) 代谢组学上游分析原理——基于 Compound Discoverer 与 Xcms 软件;
(4) Xcms 软件数据转换、提峰、峰对齐与搜库;
第二天下午
B3 R 软件基础
(1) R 和 Rstudio 的安装;
(2) Rstudio 的界面配置;
(3) R 中的基础运算和统计计算;
(4) R 中的包:包,函数与参数的使用;
(5) R 语言语法,数据类型与数据结构;
(6) R 基础画图;
B4 R 语言画图利器——ggplot2 包
(1) ggplot2 简介
(2) ggplot2 的画图哲学;
(3) ggplot2 的配色系统;
(4) ggplot2 数据挖掘与作图实战;
第三天上午
机器学习
C1 有监督式机器学习在代谢组学数据处理中的应用
(1) 人工智能、机器学习、深度学习的关系;
(2) 回归算法:从线性回归、Logistic 回归与 Cox 回归讲起;
(3) PLS-DA 算法:PCA 降维后没有差异的数据还有救吗?
(4) VIP score 的意义及选择;
(5) 分类算法:决策树,随机森林和贝叶斯网络模型;
C2 一组代谢组学数据的分类算法实现的 R 演练
(1) 数据解读;
(2) 演练与操作;
第三天下午
C3 无监督式机器学习在代谢组学数据处理中的应用
(1) 大数据处理中的降维;
(2) PCA 分析作图;
(3) 三种常见的聚类分析:K-means、层次分析与 SOM
(4) 热图和 hcluster 图的 R 语言实现;
C4 一组代谢组学数据的降维与聚类分析的 R 演练
(1) 数据解析;
(2) 演练与操作;
第四天上午
D1 在线代谢组分析网页 Metaboanalyst 操作
(1) 用 R 将数据清洗成网页需要的格式;
(2) 独立组、配对组和多组的数据格式问题;
(3) Metaboanalyst 中的上游分析(原始数据峰提取、峰对齐与搜库)
(4) Metaboanalyst 的 pipeline 以及参数设置和注意事项;
(5) Metaboanalyst 的结果查看和导出;
(6) Metaboanalyst 的数据编辑;
(7) 全流程演练与操作。
(8) 代谢联合多组学分析网页操作。
第四天下午
D2 代谢组学数据清洗与 R 语言进阶
(1) 代谢组学中的 t、fold-change 和响应值;
(2) 数据清洗流程;
(3) R 语言 tidyverse;
(4) 数据预处理:数据过滤与数据标准化(样本的 Normalization 和代谢物的 Scaling);
(5) 代谢组学数据清洗演练;
第五天上午
E1 文献数据分析部分复现(1 篇)
(1) 文献深度解读;
(2) 实操:从原始数据下载到图片复现;
(3) 学员实操。
第五天下午
E2 机器学习与代谢组学顶刊解读(3 篇);
(1) Signal Transduction and Targeted Therapy 一篇有关饥饿对不同脑区代谢组学影响变
化的小鼠脑组织代谢图谱类的文献;(数据库型)
(2) Cell 一篇代谢组学孕妇全程血液代谢组学分析得出对孕周和孕产期预测的代谢标志物
的文献;(生物标志物型)
(3) Nature 一篇对胰腺癌患者肠道菌群的代谢组学分析找到可以提高化疗效果的代谢物的
文献。(机制研究型)


深度学习在多组学融合中应用

课程报名费用:
AI蛋白质设计、AI+基因编辑、AI抗体设计:
公费价:每人每班¥6380元 (含报名费、培训费、资料费、提供课后全程回放资料)
自费价:每人每班¥6080元 (含报名费、培训费、资料费、提供课后全程回放资料)
深度学习在多组学融合中应用直播课,合成生物学与基因线路设计直播课,机器学习代谢组学:
公费价:每人每班¥5280元 (含报名费、培训费、资料费、提供课后全程回放资料)
自费价:每人每班¥4980元 (含报名费、培训费、资料费、提供课后全程回放资料)

优惠1:报二送一(同时报名两个班赠送一个学习班,赠送班任选)
两班同报:10880元
三班同报:14880元
四班同报:18880元
特惠一:24880元(可免费学习一整年本单位举办的任意课程)
特惠二:28880元(可免费学习两整年本单位举办的任意课程)
特惠三:58880元(可终身参加本单位举办的任意课程)
优惠2:提前报名缴费可享受300元优惠(仅限前15名)
优惠3:报名直播课程可赠送往期课程回放
(报名一个直播课可以赠送两个回放)
(报名三个直播课赠送下面全部课程回放)
(可点击跳转详情链接):
回放五: 本课程为视频课!CRISPR-Cas9基因编辑培训!
回放六:本课程为视频课!蛋白质晶体结构解析培训!

1、课程特色–全面的课程技术应用、原理流程、实例联系全贯穿
2、学习模式–理论知识与上机操作相结合,让零基础学员快速熟练掌握 3、课程服务答疑–主讲老师将为您实际工作中遇到的问题提供专业解答
授课方式:通过腾讯会议线上直播,理论+实操的授课模式,老师手把手带着操作,从零基础开始讲解,电子PPT和教程开课前一周提前发送给学员,所有培训使用软件都会发送给学员,有什么疑问采取开麦共享屏幕和微信群解疑,学员和老师交流、学员与学员交流,培训完毕后老师长期解疑,培训群不解散,往期培训学员对于培训质量和授课方式一致评价极高!

学员对于培训给予高度评价
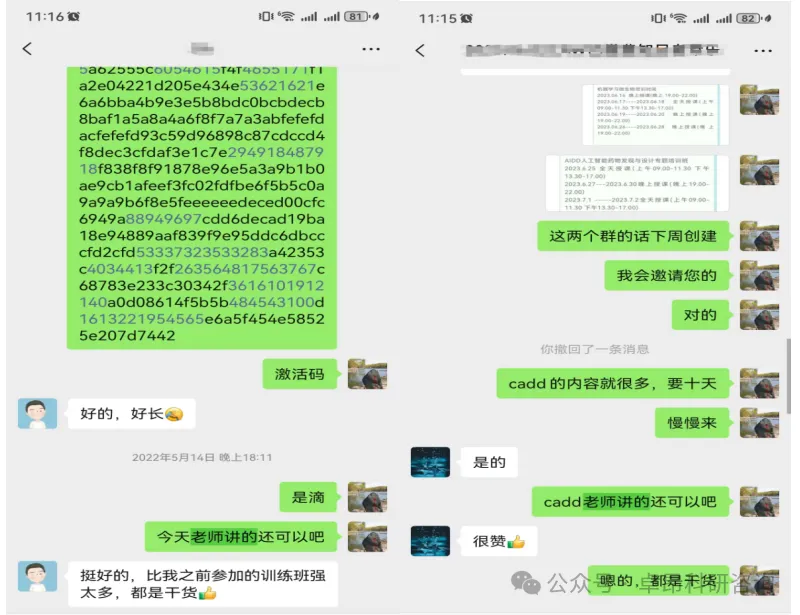
微信:Z13283822597
邮箱:m13283822597@163.com
报名电话:13283822597