 夜雨聆风
夜雨聆风





arXiv | 上海AI lab新作:ViraHinter – 双模态 AI 框架重塑病毒-宿主相互作用预测图谱
导读
在与病毒的长期“猫鼠游戏”中,病毒通过极其精简的基因组编码少量的蛋白质,却能通过与宿主蛋白质之间复杂的相互作用(vhPPIs),精准地“劫持”宿主的细胞信号、免疫防御和代谢通路。理解这些相互作用是破解病毒感染机制、寻找广谱抗病毒靶点的关键。然而,传统的实验筛选方法(如 AP-MS 或邻近标记)成本高昂、耗时巨大,且难以捕捉瞬时交互,导致目前绝大多数病毒-宿主互作组仍是未知的“暗物质”。
近日,来自上海人工智能实验室、复旦大学、上海交通大学及悉尼大学等机构的研究团队在预印本平台 arXiv 上发表了题为 “ViraHinter: a dual-modal artificial intelligence framework for predicting virus-host interactions” 的研究成果。该研究提出了一种名为 ViraHinter 的双模态深度学习框架,通过整合“序列表示”与“结构生成”两种模态,实现了对病毒-宿主相互作用的高精度预测和大规模推断,为广谱抗病毒药物开发提供了全新的数字化蓝图。
1. 现状挑战:病毒进化与数据稀疏的博弈
预测病毒与宿主蛋白相互作用面临三大难题:
-
1. 实验覆盖率极低:目前的互作数据库主要集中在少数模式病毒上,新兴病原体的互作图谱严重缺失。 -
2. 序列同源性低:病毒蛋白演化极快,基于同源性的传统方法(如序列比对)在面对跨家族的病毒时往往失效。 -
3. 空间约束缺失:早期的 AI 模型多依赖于蛋白质序列,无法捕捉互作界面的精确几何结构和物理约束。
虽然像 AlphaFold-Multimer 这样的模型在蛋白质复合物结构预测上取得了成功,但对于全蛋白质组规模的筛选来说,其计算成本过于惊人。因此,科学界迫切需要一种既能保持物理精度,又能进行高效规模化筛选的计算框架。
2. ViraHinter 架构:序列与结构的“双重奏”
ViraHinter 的核心创新在于其双模态融合设计。它不只是简单地读取序列,而是同时模拟了蛋白质的几何形态。
-
• 结构生成分支(Structure-generation branch):利用 Pairformer 模块(48层)处理输入对,通过迭代扩散(Diffusion)模块生成全原子的病毒-宿主复合物结构。这一过程提取了精确的几何特征。 -
• 序列表示分支(Sequence-representation branch):利用先进的蛋白质语言模型(如 ESM)提取嵌入向量(Embeddings),捕捉序列间的潜在关系知识。 -
• 双模态融合:研究团队设计了一个巧妙的特征聚合层,将结构分支导出的配对特征与序列分支的语义特征结合。为了防止模型仅依赖于单体折叠信号,结构特征在聚合前经过了掩码处理,强迫模型专注于“链间兼容性”。
这种设计让 ViraHinter 能够“左右开弓”:在结构证据清晰时利用空间约束;在面对演化过快、结构不稳定的病毒蛋白时,则依赖序列语义进行推断。
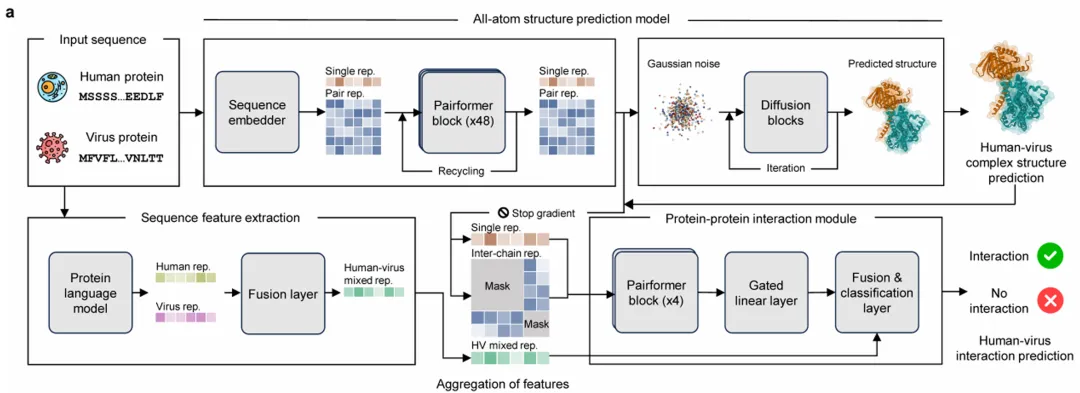
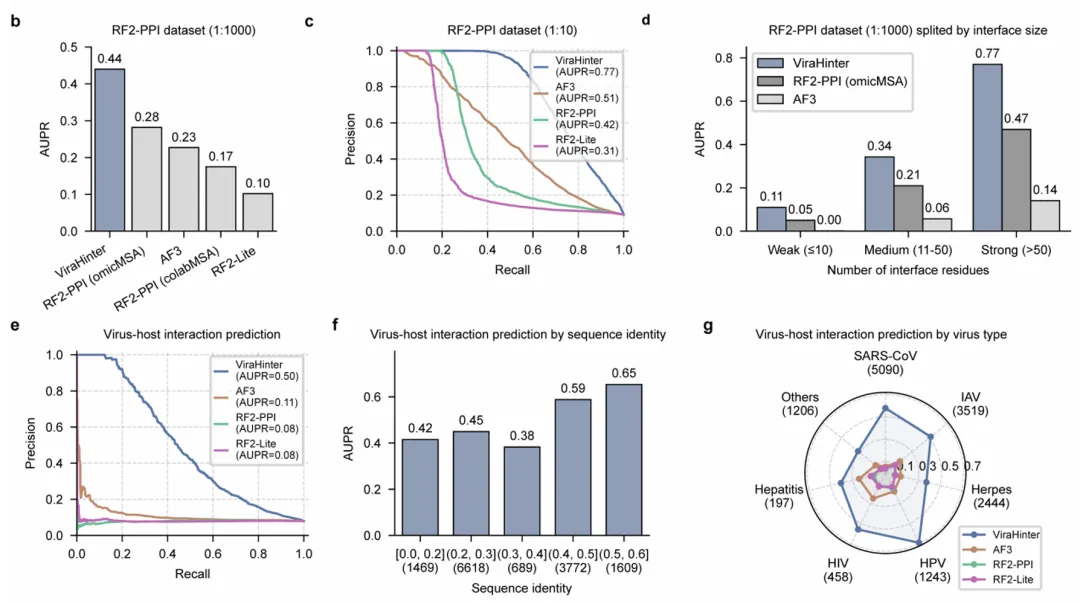
Figure 2:展示 ViraHinter 的整体架构流程图
3. 构建高保真度的病毒-宿主互作图谱
为了训练这一强大模型,研究团队首先建立了一个高质量的“金标准”数据集。他们整合了 IntAct、BioGRID、VirHostNet 和 VirusMentha 四大主流数据库,并制定了严苛的证据过滤标准:
-
• 仅保留物理关联(Physical Association),剔除共定位或遗传相互作用。 -
• 根据不同数据库的特性(如 MI 分值、实验手段)将数据分为“高置信度”和“中置信度”。
最终,基准数据集涵盖了 700 种病毒蛋白和 4,202 种人类蛋白的 10,451 个 PPI 对。通过 UpSet 统计图发现,这些数据库之间重叠极小,体现了多源整合的必要性。
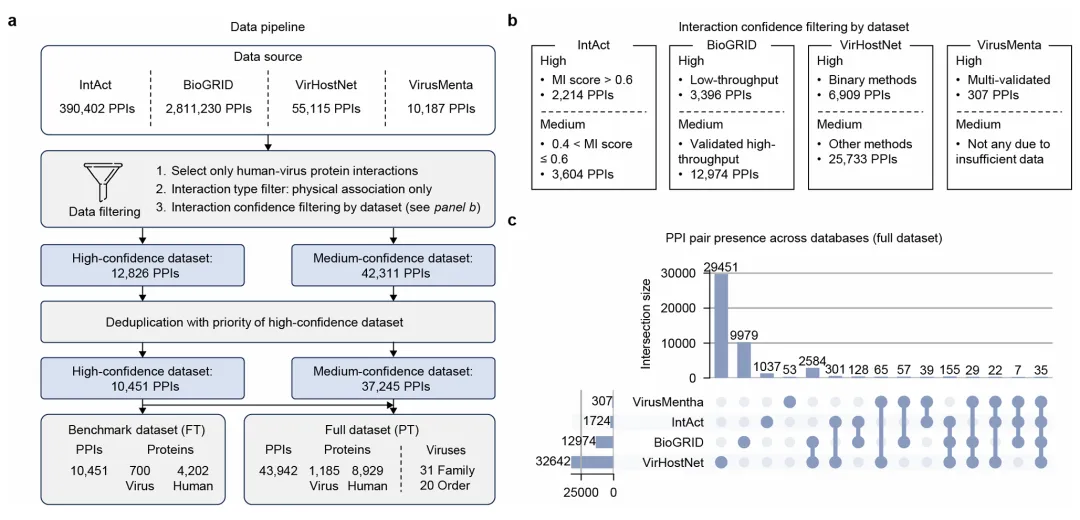
Figure 1:展示数据处理流程及各数据库之间的重叠情况(UpSet plot)
4. 性能对决:ViraHinter 遥遥领先
研究人员在极其严苛的条件下测试了 ViraHinter 的表现,特别是在 1:1,000 的极高正负样本比例下(模拟真实世界的全蛋白质组筛选场景)。
-
• 全面超越 SOTA:ViraHinter 的 AUPR 达到 0.44,显著优于 RoseTTAFold2-PPI (0.28)、AlphaFold 3 (0.23) 和 RoseTTAFold2-Lite (0.10)。 -
• 跨越“同源性陷阱”:在“病毒留出”(Virus-held-out)测试中,即使测试集的病毒与训练集序列相似度不足 20%,ViraHinter 依然保持了强大的预测能力,其表现比 AlphaFold 3 高出约 4.5 倍。 -
• 全界面覆盖:无论是大而稳定的相互作用界面,还是只有不到 10 个残基的弱交互界面,ViraHinter 均表现稳健。
5. 实战案例一:解析冠状病毒的“秘密联系”
针对 SARS-CoV-2、SARS-CoV-1 和 MERS-CoV 三种致命冠状病毒,ViraHinter 对 958 对候选 PPI 进行了验证。
-
• 精准排名:模型给出的得分与实验证据强度呈正相关。在高置信度区间(Top 1%),ViraHinter 对已知互作蛋白的富集度达到了随机预测的 4 倍。 -
• 发现泛冠状病毒靶点:ViraHinter 预测了 8 个关键宿主因子,如 RAB8A、RAB5C、PABPC1 等。这些因子被预测能与所有三种冠状病毒的多种非结构蛋白(如 NSP7)结合。 -
• 结构守恒性:尽管三种病毒的 NSP7 序列存在差异,ViraHinter 预测其与 RAB8A 的结合模式高度一致,均指向同一个功能口袋,暗示了这些互作轴在进化上的极端重要性。
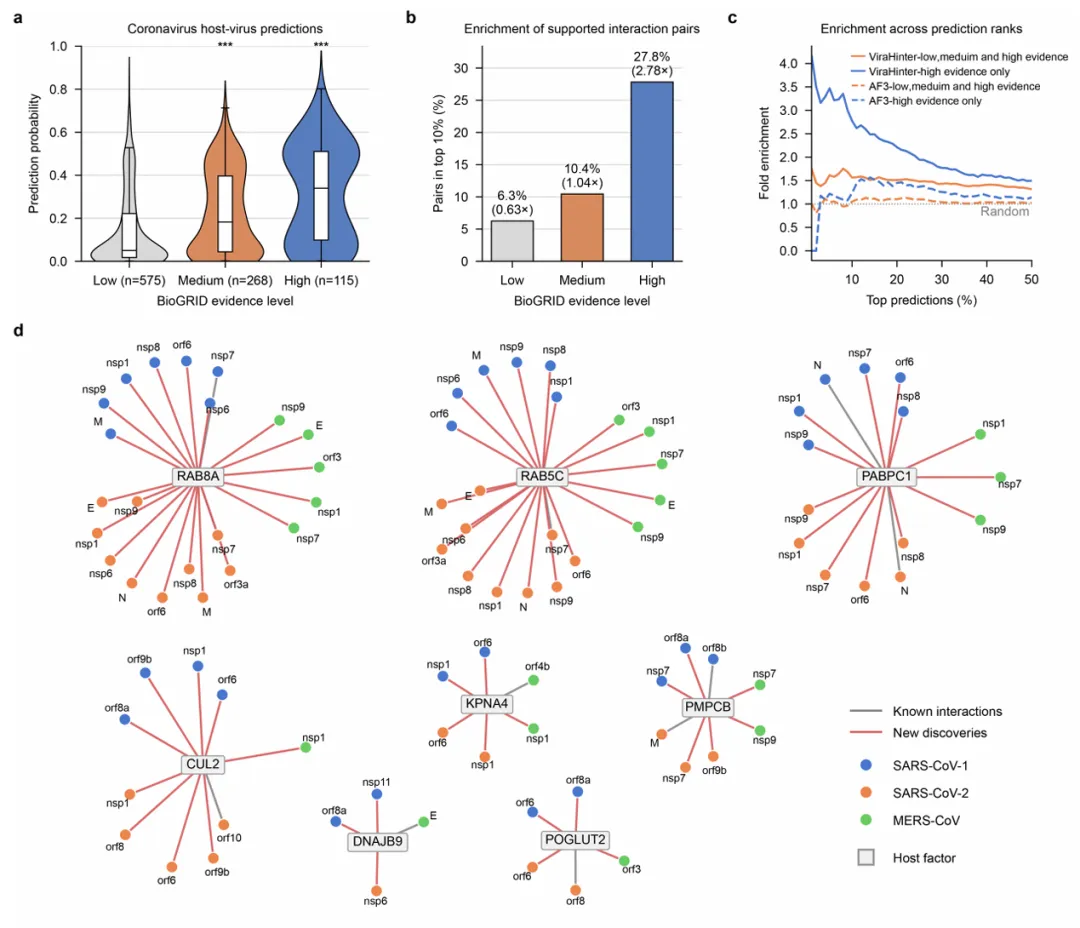
Figure 3:展示冠状病毒的预测得分分布及泛冠状病毒互作网络图
6. 实战案例二:流感病毒的“演化瓶颈”
流感病毒(IAV)具有高度的变异性。研究团队对比了 H1N1、H3N2 以及一种禽源的 H9N2 毒株,甚至使用了实验室 2025 年最新测序的 H3N2 临床株。
-
• 33 个核心宿主因子:通过取交集,ViraHinter 锁定了 33 个所有流感亚型共同依赖的“核心因子”。这些因子聚集在囊泡运输、细胞骨架、染色质与 DNA 损伤、RNA 代谢及信号传导五大功能模块中。 -
• 捕捉瞬时动态:ViraHinter 成功识别了如 NS1、M1、NP 以及聚合酶亚基(PB1/PB2/PA)在这些毒株中的共有靶点。 -
• RAB11A 的关键作用:模型预测流感的 HA 蛋白与宿主的 RAB11A 存在高度保守的相互作用,这为病毒颗粒的转运提供了分子基础。结构分析显示,这种结合界面在不同亚型中几乎重合,具有开发广谱抗病毒药物的巨大潜力。
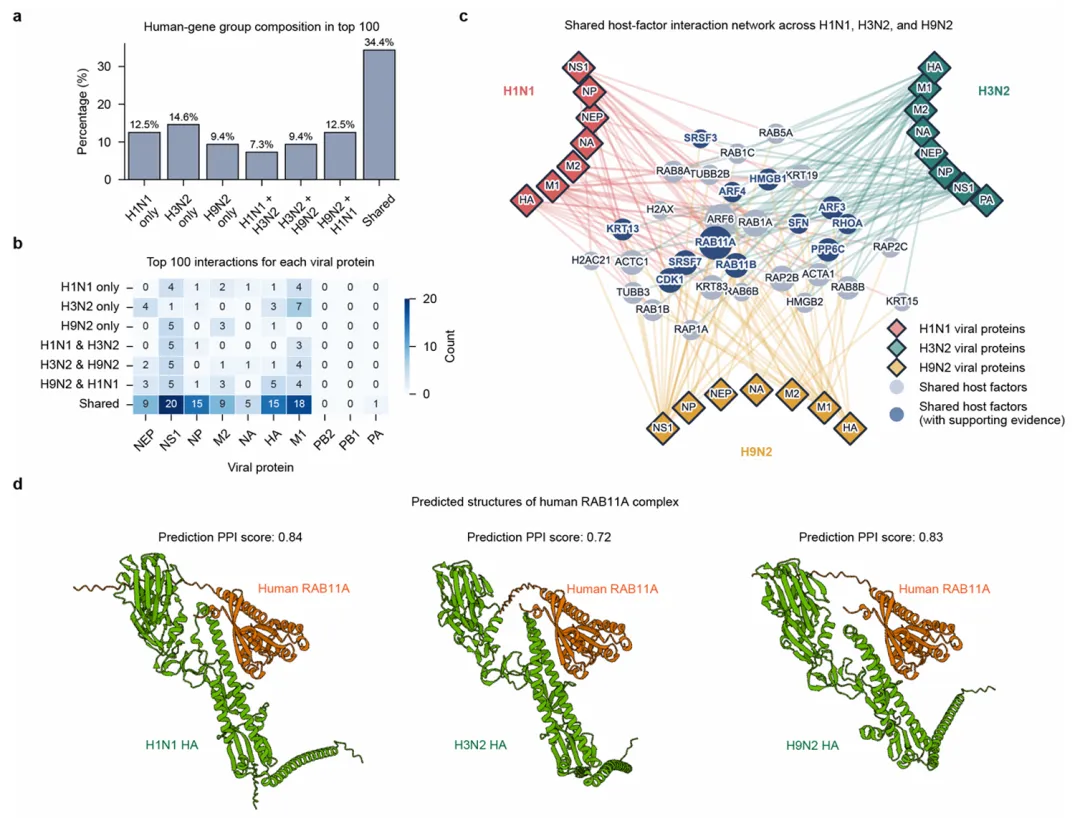
Figure 4:展示流感病毒 33 个共有因子的互作网络及 HA-RAB11A 的保守复合物结构
总结与展望
ViraHinter 的出现标志着病毒学研究进入了“预测先行”的新阶段。通过双模态 AI 技术,我们不仅能以前所未有的速度筛选潜在的病毒靶点,还能在原子分辨率下观察它们是如何相互攻击与防御的。
正如论文通讯作者 Weifeng Shi 和 Siqi Sun 所言,ViraHinter 为所有已知的人类感染病毒提供了系统性筛查宿主因子的能力,这不仅能加速新型疗法的发现,更为应对未来可能出现的“病毒 X”(Disease X)储备了关键的计算手段。
参考文献:Bai et al., “ViraHinter: a dual-modal artificial intelligence framework for predicting virus-host interactions.” arXiv (2026).