 夜雨聆风
夜雨聆风





荧光定量PCR引物设计避坑指南:从序列下载到异常排查
荧光定量PCR引物设计避坑指南:从序列下载到异常排查

荧光定量PCR(qPCR)是应用最广、效率最高的检测方法。但许多实验室技术人员都曾遇到这样的窘境:病料典型、提取规范、试剂正常,可就是跑不出理想的扩增曲线;或者熔解曲线出现双峰,阴性对照莫名其妙起峰……反复折腾后才发现,问题的根源往往出在最基础却最容易被轻视的环节——引物设计。
引物设计不是“找个保守区,往软件里一输就完事”那么简单。比如H9N2病毒的HA基因不断变异,数据库里已收录成千上万条序列;不同流行株之间可能存在关键位点差异,若引物设计时覆盖的毒株代表性不足,轻则检测灵敏度下降,重则出现假阴性,漏掉真正的阳性样本。此外,引物自身二级结构、二聚体、GC含量、Tm值匹配度等参数稍有偏差,就可能导致非特异性扩增或扩增效率低下。
那么,如何从零开始设计一套特异、灵敏、稳定的qPCR引物?从靶基因选择、序列下载与比对,到保守区定位、引物参数优化,再到软件验证和常见异常排查。以H9N2进行举例:
一、设计前的准备工作
1. 选择靶基因:HA基因(最推荐)
H9N2亚型禽流感病毒的血凝素(Hemagglutinin, HA)基因是该亚型特异性最强的标志基因,也是检测方法的首选靶标。选择HA基因是因为它具备以下三个关键优势:
亚型特异性高:HA基因是决定病毒亚型的关键基因,不同亚型(H1-H18)之间差异显著,选择HA基因设计引物可以专门针对H9亚型。
序列资源丰富:GenBank数据库中收录了大量来自不同地区、不同年份、不同宿主来源的H9N2分离株的HA序列,为保守区域筛选提供了充足的数据支持。
突变热点区域明确:HA基因虽然整体存在变异,但其跨膜区和部分茎部区域相对保守。通过多序列比对可以准确定位保守区段,确保引物能够覆盖不同流行株。
文献依据:目前国内已有多项H9N2荧光定量PCR检测方法研究,均采用HA基因保守区域作为引物设计靶位点,验证表明该方法灵敏度可达0.1 copies/μL,特异性良好且与其他禽病原无交叉反应。
2. 序列获取:有多少序列才够”代表性”?
最低下载量建议(分级方案):
1)快速方案(适用于紧急检测需求):下载100-200条H9-HA代表性序列,包括我国周边和国内近5年流行的主要毒株;
2)标准方案(适用于常规方法建立):下载NCBI所有H9-HA序列,同时挑选H1-H14的HA代表株进行比对;
3)最佳方案(适用于学术研究和精确检测):下载NCBI所有H9-HA序列及其他分型的流感HA序列,同时结合本地测序H9流行株序列,涵盖尽量多的时间跨度和地域来源进行引物设计,同时定期使用最新分离株进行引物复核,保证检测准确性。
3. 序列来源与下载策略
|
来源 |
用途 |
注意事项 |
|
NCBI GenBank |
下载全球参考毒株和已发表序列 |
优先选择”complete cds”(完整编码序列)条目,确保序列质量 |
|
GISAID(全球流感数据共享倡议平台) |
获取最新流行株序列 |
需要申请账号,数据更新更快,包含实时监测数据 |
|
内部测序数据 |
补充本地流行株信息 |
对于区域性检测方法建立尤为重要 |
序列筛选要点:下载时优先选择近5年的分离株、覆盖中国各省份及周边国家(如韩国、日本、越南等)的毒株、来源于商品肉鸡、蛋鸡、水禽及野生鸟类的不同宿主来源分离株。
4. 序列比对与保守区筛选
4.1 比对软件选择
获取序列后,需要使用多序列比对软件进行对齐和保守性分析:
|
软件 |
操作方式 |
适用场景 |
|
DNAman |
图形化界面操作 |
适合批量序列比对和保守区域可视化分析- |
|
MEGA 7.0/11.0 |
图形化界面操作 |
经典的分子进化遗传学分析工具,适合序列比对和保守性分析 |
|
CLC Genomics Workbench |
图形化界面操作 |
适用于大规模序列数据处理和可视化分析 |
|
MAFFT |
在线/命令行 |
对大规模序列比对速度快、精度高 |
|
ClustalW/Clustal Omega |
在线/命令行 |
经典比对工具,Omega适合大序列集 |
|
BioEdit |
图形化界面 |
序列编辑和保守性可视化分析 |
4.2 保守区域识别:三个高级操作技巧
在比对结果中标记出所有序列完全相同的区域(100%保守),以及多数序列相同的区域(>90%保守)。
① 利用”序列一致性矩阵”精准定位:大多数比对软件(如MEGA、BioEdit、CLC)都能生成序列一致性矩阵图表,通过颜色深浅直观显示保守程度。颜色越深表示该位置越保守,优先选择所有序列完全一致的区域设计引物。
② 使用”WebLogo”可视化保守性:将比对好的序列上传至WebLogo在线工具(https://weblogo.berkeley.edu/),生成序列Logo图。纵轴高度代表该位点的保守程度,柱子越高表示该位置越保守。这是判断保守区质量的直观方式。
③ 关注密码子第3位的”容忍性”:由于密码子简并性,DNA序列的第3位碱基突变不一定会改变氨基酸。在挑选引物位置时,若某区域在第1、2位保守(氨基酸保守),即使第3位有轻微变异仍可使用。
二、引物设计核心参数
1. 基础参数设置
|
参数 |
推荐值 |
关键考量 |
|
引物长度 |
18-25 bp |
过短特异性差,过长Tm值过高影响效率 |
|
GC含量 |
40%-60% |
极端GC含量导致二级结构增多,影响扩增稳定性 |
|
Tm值(解链温度) |
58-65℃,上下游引物ΔTm ≤ 2℃ |
Tm值过低会导致非特异性扩增;ΔTm过大会影响退火温度优化空间,导致部分引物无法有效结合模板 |
|
产物长度 |
80-250 bp(qPCR最佳80-150 bp) |
短片段扩增效率更高,适合实时定量 |
|
3′ 端 |
最后5个碱基内避免连续G/C |
避免非特异性延伸;3′ 端最后一个碱基首选G或C(但不要连续) |
|
连续碱基 |
避免>4个连续G,避免任何>4个单碱基重复 |
连续G易形成G-四链体、二级结构增多,影响引物结合效率 |
|
引物间互补 |
3′ 端避免互补对 |
引物二聚体(尤其是3’端互补)会显著降低扩增效率 |
引物二聚体的判断:估算引物二聚体的形成趋势,可以使用∆G(自由能)参数。3′ 端互补形成的二聚体∆G绝对值越大,二聚体越稳定,对反应的干扰越大。
2. 四种常用引物/探针方案的简要点评
在H9N2检测中,根据检测目的和条件选择合适的方法:
|
方案类型 |
工作流程 |
成本 |
优点 |
缺点 |
适用场景 |
|
SYBR Green I法 |
一对引物 |
低 |
操作简单,成本低廉,适合方法探索 |
特异性依赖引物质量,需后续熔解曲线验证单一产物 |
实验室初步筛查、基因表达分析 |
|
TaqMan探针法 |
一对引物+一条探针 |
较高 |
双特异性(引物+探针),背景低,可多重检测 |
探针合成成本高,设计更复杂 |
临床样品检测、病毒载量定量 |
|
MGB探针法 |
一对引物+MGB探针 |
高 |
探针更短,Tm值更稳定,信噪比更高 |
合成成本最高 |
追求高灵敏度、复杂样品背景下的检测 |
|
双重荧光PCR |
两对引物+两条探针(双色标记) |
最高 |
同时检测HA和NA两个基因,减少假阴性 |
设计难度最大,需避免引物间交叉干扰 |
临床确诊、疫苗评估 |
3. 序列位置的特殊考量
避免非保守区域:H9N2病毒的HA基因具有较高变异性,设计时务必避开HA基因的高变区(特别是受体结合域及其周边区域);
优选跨膜区/茎部区:HA基因的跨膜区和茎部区相对保守,是引物设计的优选位置;
避免3′ 端单核苷酸重复:超过4个连续碱基重复(如AAAAA)会导致滑移错配。
三、使用Primer-BLAST软件设计与特异性验证
Primer-BLAST是一个集引物设计与特异性验证于一体的免费在线工具。在CLC Genomics Workbench中完成了序列导入和整理,可以直接导出FASTA格式序列,或者直接复制选择的重合度较高的区域序列,再配合Primer-BLAST完成引物设计。
1. 操作流程
进入Primer-BLAST:访问https://www.ncbi.nlm.nih.gov/tools/primer-blast/
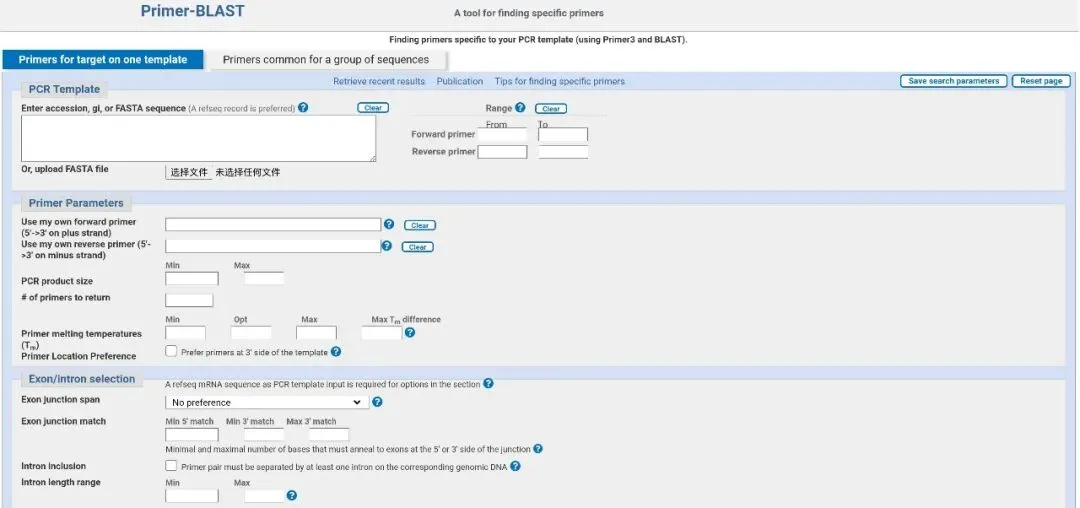
粘贴序列:将您的比对保守区域序列粘贴到”PCR Template”框中。也可以直接输入GenBank登录号,或粘贴FASTA格式的序列。
设置引物参数:按上述核心参数填写。
选择数据库和物种:Database选择”nr”,Organism输入”Avian influenza virus H9N2″。
调整Blast搜索参数:Primer specificity stringency设置为3-4,即允许多个错配。
运行并手动筛选:避开同源区域、重复序列和已知变异位点。
2. 特异性验证的判定标准
理想状态:所有匹配均落在H9N2的HA基因内,且靶向区域在所有H9流行株中完全保守,与其他亚型病毒基因组无同源性。
临界状态:若与其他亚型存在≤2个连续的错配碱基或≤3个分散错配,可在退火温度优化中通过区分。
不可接受:完全匹配的扩增子出现在H9N2以外基因,或其他禽类病原体中出现明显的同源性(连续错配数<3),需更换引物。
3. NCBI查找保守序列的可视化网站补充
除了Primer-BLAST和序列比对软件外,以下在线工具也值得使用:
NCBI Conserved Domain Database (CDD):输入序列,自动标识HA基因的保守结构域区域。
Influenza Virus Database (NCBI流感病毒专用数据库):专门收录流感病毒序列,可按亚型、宿主、年份筛选,直接下载筛选后的序列集进行比对。
四、引物异常排查
常见问题优先级排序
优先级1:无扩增产物或CT值过大
原因1:模板问题——模板降解或浓度过低→ 对策:重新提取RNA,提高模板浓度,梯度稀释测试
原因2:引物降解——合成引物存放过久或反复冻融导致断裂→ 对策:PAGE电泳检查引物完整性,重新合成分装保存
原因3:退火温度过高——超出引物Tm值范围5℃以上→ 对策:以Tm值-5℃为起点进行温度梯度PCR优化
原因4:引物间或引物自身存在严重二级结构→ 对策:使用OligoAnalyzer评估二级结构,重新设计
优先级2:非特异性扩增(出现多条带或熔解曲线多峰)
原因1:退火温度过低——引物与模板非特异性结合→ 对策:设置温度梯度PCR(在Tm值±5℃范围内),或使用Touchdown PCR(每循环降低0.5-1℃)
原因2:Mg²⁺浓度过高——增强非特异性延伸活性→ 对策:优化Mg²⁺终浓度(常规1.5-2.5 mM,qPCR推荐3-5 mM)
原因3:引物浓度过高——促进引物二聚体和非特异性结合→ 对策:常规PCR引物终浓度0.2-0.5 μM,qPCR 10-50 nM
原因4:引物自身设计缺陷→ 对策:使用Primer-BLAST重新排查并设计引物
优先级3:出现引物二聚体(熔解曲线约80℃以下出现小峰)
原因1:引物3’端互补——上下游引物3’端存在互补序列→ 对策:检查3’端最后8个碱基,确保无连续2个以上互补碱基,并避免以AAAAAAAA结尾的引物序列
原因2:循环数过多(>35循环)→ 对策:控制循环数≤35,提前终止扩增
优先级4:标准曲线线性差
原因1:标准品降解——反复冻融导致降解→ 对策:分装保存,每次实验使用新鲜稀释的标准品
原因2:加样误差——标准品稀释梯度操作不准确→ 对策:使用预混液统一配制,大体积稀释减少误差
原因3:模板中抑制物——提取核酸混入残留杂质→ 对策:将模板适度稀释(5-10倍)后加入反应体系
优先级5:阴性对照出现信号
原因1:试剂污染——预混液或水被扩增产物污染→ 对策:更换新试剂,实验前后用DNA去污剂(如DNAzap)清洁操作台面和无菌操作台
原因2:引物二聚体干扰——仅在使用SYBR Green法时出现→ 对策:结合熔解曲线判定,设置无模板对照(NTC)
五、不同方法的额外注意事项
1. SYBR Green法(尤其注意)
SYBR Green荧光染料可与任何双链DNA结合,这意味着引物二聚体和非特异性扩增产物都会产生荧光信号,导致定量结果不准确。熔解曲线是验证特异性关键工具,理想熔解曲线应为单一尖峰。若出现双峰或弥散峰,应重新设计引物或优化反应条件。
2. TaqMan探针法
探针Tm值应比引物Tm值高5-10℃,确保探针优先结合模板
探针长度20-30 bp(TaqMan)或16-25 bp(MGB),5’端避免使用G碱基探针设计时,让引物尽可能靠近探针,扩增片段较短(70-150 bp)有利于探针充分水解,因为探针长度通常在20-30 bp之间,TM值较高,设计中必须确保其完全落在引物扩增区域内,且避开发夹结构和重复序列。
探针区域需额外验证保守性:探针长度通常比引物长(20-30 bp),在序列保真度上要求更高。与引物筛选相同,必须通过多序列比对确保探针靶向区域在所有流行株中完全保守,否则可能存在假阴性。



END
