夜雨聆风
夜雨聆风关键词:阿尔茨海默病,线粒体功能障碍,iPSC,Presenilin 1,CRISPR,淀粉样前体蛋白
keyword:Alzheimer's, mitochondrial dysfunction, iPSC, presenilin 1, CRISPR, amyloid precursor protein
期刊名:《International Journal of Biological Sciences》影响因子(IF):10,分区(JCR):Q1
标题:中文 APP缺陷改善家族性阿尔茨海默病早老素1 F105C和A246E突变诱导的人皮层神经元线粒体功能障碍(英文 APP Deficiency Ameliorates FAD Presenilin 1 F105C and A246E Mutations-induced Mitochondrial Dysfunction in Human Cortical Neurons)
第一作者、通信作者及单位:Yu-Hsin Yen(杜克-新加坡国立大学医学院)、Fang Yuan(杜克-新加坡国立大学医学院);通信作者:Su-Chun Zhang(桑福德伯纳姆普雷比斯医学发现研究所)、Cheong-Meng Chong(澳门大学)、Huanxing Su(澳门大学)
发表时间:2026年2月18日
主要结论概要:本研究通过分析散发性AD患者脑组织数据,结合CRISPR/Cas9基因编辑技术构建携带不同早老素1(PS1)突变的人诱导多能干细胞(iPSC)来源的皮层神经元模型。研究发现,PS1突变会导致神经元内淀粉样前体蛋白(APP)在线粒体中积累,引发线粒体功能严重障碍及AD相关病理。关键的是,敲除APP能够显著改善这些线粒体缺陷,并减轻tau蛋白过度磷酸化和Aβ产生,揭示了APP在介导PS1突变所致线粒体功能障碍中的核心作用。
线粒体功能障碍被认为是阿尔茨海默病(AD)早期且核心的病理特征之一。过往研究提示,淀粉样前体蛋白(APP)在线粒体内的积累可能与此有关,而作为家族性AD最常见病因的早老素1(PS1)突变也被证明会损害线粒体功能[1]。然而,APP是否以及如何影响PS1突变诱导的线粒体缺陷,其具体机制在人神经元中尚未明确。近期发表于《International Journal of Biological Sciences》的研究[1] 利用前沿的干细胞与基因编辑技术,深入探究了这一问题,为理解AD发病机制提供了新的重要视角。
散发性AD脑组织与PS1突变神经元均呈现线粒体异常
研究团队首先分析了来自Mount Sinai脑库的散发性AD患者死后脑组织的转录组和蛋白质组数据。他们发现,在内嗅皮层(Brodmann 36区),与线粒体功能相关的通路,特别是氧化磷酸化和呼吸链复合物相关基因与蛋白的表达发生了显著改变[1]。这提示在患者大脑中,线粒体功能已广泛受损。为了在更可控的系统中验证并探索其机制,研究人员利用CRISPR/Cas9技术构建了两对同基因型的人iPSC系:一对是在健康iPSC中引入PS1 F105C突变及其野生型对照;另一对是将患者来源的携带PS1 A246E突变的iPSC进行基因校正,获得同基因野生型对照[1]。将这些iPSC分化为皮层神经元后,与对照组相比,两种PS1突变神经元均出现了典型的AD病理表型,包括Aβ40和Aβ42分泌增加,以及tau蛋白的过度磷酸化[1]。

图1(Figure 1):PS1突变神经元中线粒体形态与功能异常
图1-a和b显示,与对照组神经元中线粒体(绿色TOM20)呈长管状分布不同,PS1 F105C和A246E突变神经元中的线粒体变得短小、碎片化。图1-c定量分析证实突变神经元平均线粒体长度显著缩短。图1-d和e的TMRE染色显示,突变神经元的线粒体膜电位显著降低,表明其功能受损。
进一步的功能检测证实了严重的线粒体功能障碍。PS1突变神经元中的线粒体变得短小、碎片化,线粒体膜电位显著下降。同时,负责线粒体分裂的蛋白DRP1和FIS1表达上调,而呼吸链复合物关键蛋白(如NDUFB8、UQCRC2、COX II)的表达则降低[1]。这些发现与在散发性AD脑组织中的观察一致,表明PS1突变足以在人皮层神经元中直接诱发线粒体结构和功能的双重损害。
APP在线粒体异常积累是导致功能障碍的关键环节
一个关键的发现是,APP在PS1突变神经元的线粒体中出现了异常积累。通过超高分辨率显微镜观察,研究人员发现PS1突变神经元中,APP蛋白与线粒体外膜蛋白TOM20的共定位显著多于对照组[1]。进一步的生化实验表明,APP通过其N端的线粒体靶向序列与TOM20结合,从而定位于线粒体外膜。同时,PS1突变神经元中全长APP的表达水平也显著升高[1]。这些结果将PS1突变、APP表达与定位异常、以及线粒体功能障碍直接联系起来,提示APP可能是PS1突变损害线粒体的重要媒介。
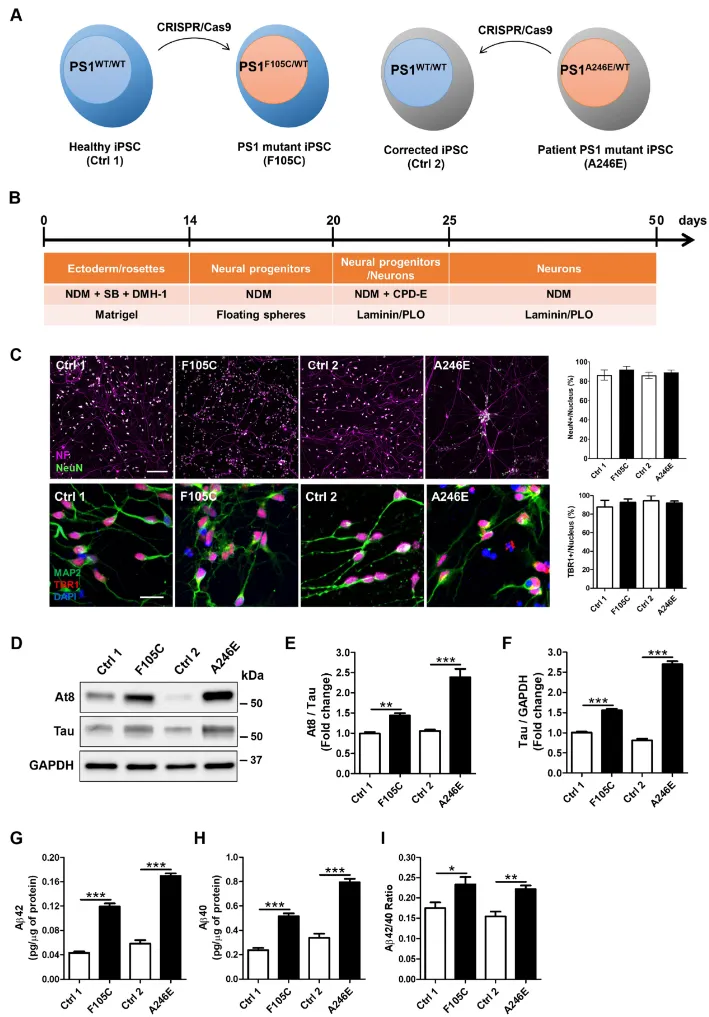
图2(Figure 2):敲除APP逆转PS1突变神经元的线粒体与病理缺陷
图2-a和b显示,在PS1 F105C突变神经元中敲除APP(F105C_APPKO)后,原本碎片化的线粒体(绿色)恢复为类似对照组的长管状形态。图2-c定量显示APP敲除后线粒体膜电位得到显著提升。图2-d的蛋白质印迹分析表明,APP敲除后,tau蛋白的磷酸化水平(AT8/tau比值)以及线粒体分裂蛋白DRP1的表达均降低,而线粒体呼吸链复合物蛋白(I-IV)的表达则有所恢复。
敲除APP可显著改善线粒体功能与AD病理
为了直接验证APP的作用,研究团队在PS1 F105C突变的iPSC中进一步敲除了APP基因。令人振奋的是,这一操作产生了显著的“挽救”效果。与未敲除APP的PS1 F105C突变神经元相比,APP敲除的神经元其线粒体形态恢复为正常的长管状,线粒体膜电位显著回升[1]。与此同时,线粒体呼吸链复合物蛋白的表达水平得到恢复,分裂蛋白DRP1的表达下降。更为重要的是,AD相关的病理表型也得到改善:tau蛋白的过度磷酸化水平降低,且细胞不再分泌Aβ[1]。在海马分析实验中,APP敲除也提升了神经元的基础呼吸和ATP产量[1]。这些证据强有力地表明,APP是PS1突变诱发线粒体功能障碍和部分AD病理变化的下游关键效应分子。
这项研究巧妙地结合了患者组织数据与精确基因编辑的iPSC神经元模型,首次系统地证明了在家族性AD相关的PS1突变背景下,APP的异常表达与线粒体定位是导致神经元线粒体功能缺陷的核心环节。这为AD,特别是由PS1突变驱动的AD,提出了一个超越传统Aβ瀑布假说的新机制框架:PS1突变 → APP表达增加/线粒体定位 → 线粒体功能障碍 → 神经元变性与AD病理[1]。该发现不仅加深了对AD复杂发病网络的理解,也为开发新的治疗策略指明了方向,即靶向APP的线粒体转运或其与线粒体的相互作用,可能成为干预AD早期病理过程的有效手段。
参考文献
[1] Yen, Yu-Hsin et al. “APP Deficiency Ameliorates FAD Presenilin 1 F105C and A246E Mutations-induced Mitochondrial Dysfunction in Human Cortical Neurons.” International journal of biological sciences vol. 22,5 2720-2735. 18 Feb. 2026. https://doi.org/10.7150/ijbs.120062
本文内容来自公开信息及研究文献,不代表临床建议。
如需投稿、转载、商务合作后台留言,回复有一定延迟敬请谅解。
辅助编辑:腾讯元宝or(GPT、Gemini、Claude等)
扫码加入ima知识库,获取本公众号所有文献全文资料,AI问答了解更多信息,文献实时更新