做分子动力学(MD)模拟的朋友,基本都离不开LAMMPS。但计算跑完后,面对一大堆 dump轨迹文件:结构怎么看?运动过程怎么展示?文章里的高清分子图怎么画?这时候就必须要请出今天的主角 ——VMD(Visual Molecular Dynamics)。
什么是 VMD?
VMD 是由美国伊利诺伊大学厄巴纳-香香分校(UIUC)开发的免费、开源分子可视化与分析软件。它不仅能高质量渲染蛋白质、核酸、纳米材料等体系,还广泛用于 LAMMPS / GROMACS / NAMD 等 MD 软件的后处理:一句话总结:LAMMPS 负责算,VMD 负责“让你看见”和“把结果讲清楚”。
🌐 官网与下载
http://www.ks.uiuc.edu/Research/vmd/https://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD如不想注册,可后台回复VMD获取百度网盘下载链接。推荐选择 1.9.4 稳定版;alpha 版适合想尝鲜的用户。
💻 Windows 下安装 VMD
进入官网下载页,选择Windows 64-bit(如 vmd-1.9.4a53.exe)Next → I Agree → Install(可以自己改路径) → Finish安装完成后,桌面出现 VMD 图标,打开后一般可见:
🐧 Ubuntu (Linux) 下安装 VMD
以 vmd-1.9.4为例(文件名按你实际下载的来):1. 解压
tar -xvzf vmd-1.9.4.bin.LINUXAMD64.tar.gz2. 配置
3. 编译安装
▶️ 简单运行:加载 LAMMPS 轨迹
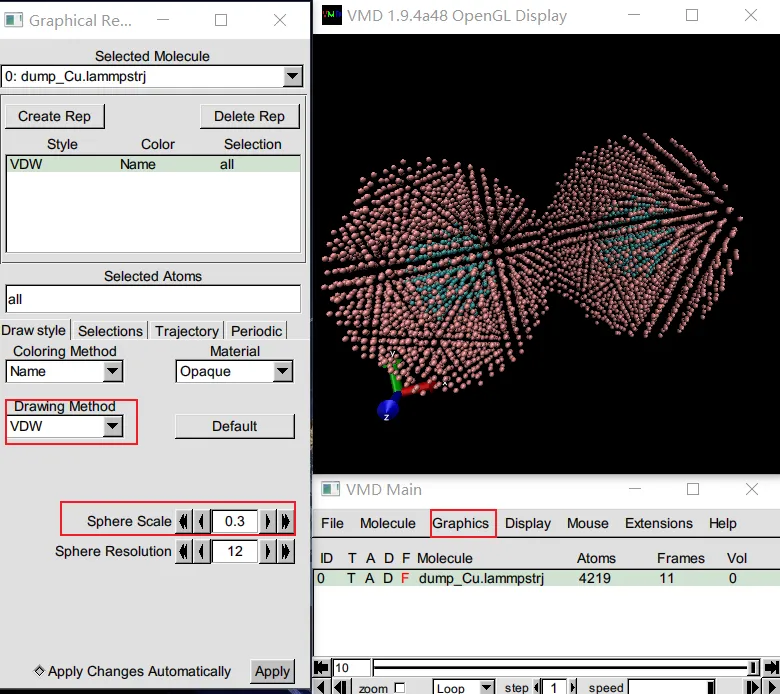
打开 VMD → 菜单File → New Molecule同一窗口再次Browse,选择轨迹文件【注意,文件路径不要有中文,否则容易报错,无法读入】(如 dump.lammpstrj)→LoadGraphics → Representations
📌 小结
对LAMMPS 用户尤其友好,原生支持 LAMMPS 轨迹跨平台(Windows / Linux / macOS),免费,插件生态丰富能做图、做动画、做分析,几乎“科研展示三件套”都能覆盖
如果你觉得这篇教程有用,欢迎点赞 + 在看 + 收藏📌也欢迎关注本公众号,后台回复「VMD」获取我整理好的:后台回复关键词:VMD
 夜雨聆风
夜雨聆风