 夜雨聆风
夜雨聆风下载说明
[软件名称]:Modifit
[界面语言]:英文
[软件分类]:生物医学
本文章长期有效,请在文末获取最新下载链接。
软件介绍
Modifit 是一款功能十分全面的分子模拟软件。
Modifit 3.1 是一款专为分子动力学模拟与结构优化设计的专业科研软件,广泛应用于生物大分子、有机材料及纳米体系的结构修饰与构象分析。该版本由计算化学团队基于多年算法积累开发,核心目标在于解决传统分子模拟中力场参数不匹配、局部极小值陷阱及大体系收敛缓慢等痛点。Modifit 3.1 集成了最新的混合蒙特卡罗与梯度下降耦合算法,能够在保持量子力学精度的前提下,对含数万原子的复杂体系进行快速结构弛豫。其内置的自动势函数校正模块,可针对非标准残基或配体分子动态调整力场参数,显著提升模拟结果的可靠性。无论您是研究蛋白质药物结合构象,还是设计新型聚合物材料,Modifit 3.1 都能提供从初始模型构建到最终结构验证的一站式解决方案。版本特点:1. 多尺度力场融合引擎:支持 CHARMM36、AMBER ff19SB、OPLSAA 及自定义力场的无缝切换与混合使用,可在同一模拟中同时处理蛋白、核酸与小分子配体。2. 自适应退火优化:引入模拟退火与遗传算法结合的全局搜索策略,自动跳出局部能量极小值,特别适用于柔性大分子或表面吸附体系的构象扫描。3. 实时静电修正:基于泊松玻尔兹曼隐式溶剂模型,在结构优化过程中动态计算溶剂化效应,避免真空模拟导致的构象失真。4. 并行加速与断点续算:支持 CPU/GPU 混合并行,单卡 RTX 4090 即可处理 10 万原子体系;模拟过程每 5 分钟自动保存 checkpoint,防止意外中断造成数据丢失。5. 内置验证工具包:自动生成拉氏图、RMSD 轨迹图及能量分解报告,无需额外脚本即可评估优化结果的物理合理性。支持格式:导入格式:.pdb(蛋白质数据库文件)、.mol2(Tripos 分子格式)、.sdf(结构数据文件)、.gro(GROMACS 坐标格式)、.psf(CHARMM 拓扑文件)、.xyz(通用笛卡尔坐标)、.cif(晶体学信息文件)。导出格式:.pdb、.gro、.xyz、.mol2、.dcd(轨迹文件)、.trr(GROMACS 全精度轨迹)、.crd(AMBER 坐标)、.prmtop(AMBER 拓扑)、.psf、.itp(GROMACS 拓扑)。附加格式:.rtf(残基拓扑)、.par(力场参数文件)、.log(能量与收敛日志)、.csv(统计表格)。使用流程:第一步:启动 Modifit 3.1,点击“导入结构”按钮,选择待处理的 .pdb 或 .mol2 文件,系统自动解析原子类型并提示缺失残基或异常键长。第二步:在“力场设置”面板中选择目标力场(如 AMBER ff19SB),若体系包含非标准分子,需手动加载 .rtf 和 .par 文件,或使用自动参数分配功能。第三步:进入“优化策略”选项卡,设定溶剂模型(如隐式水或显式水盒)、温度(300K)及迭代步数(默认 5000 步),勾选“自适应退火”以增强全局搜索能力。第四步:点击“运行”启动计算,实时监控能量收敛曲线与 RMSD 波动;若出现震荡,可暂停并调整步长或温度参数。第五步:优化完成后,在“结果分析”界面查看最终构象,导出优化后结构为 .pdb 文件,并保存能量分解报告与轨迹文件用于后续可视化。软件安装
1. 文件解压后会产生多个文件,这些文件不要删除,打开下图箭头所指的图标
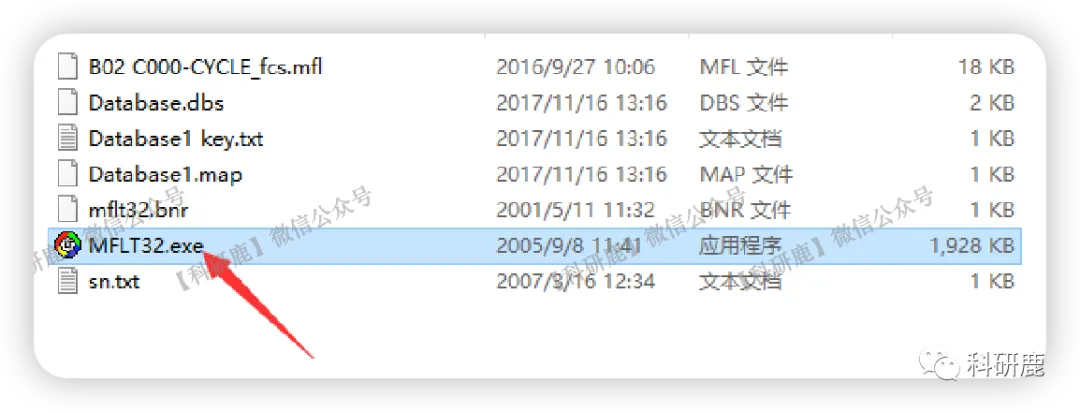
2. 打开上述图标会弹出一系列弹窗,点击“确定”或“OK”。
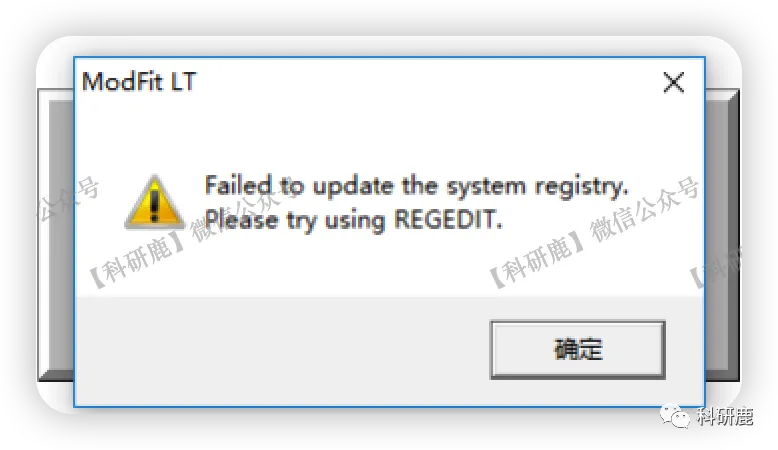
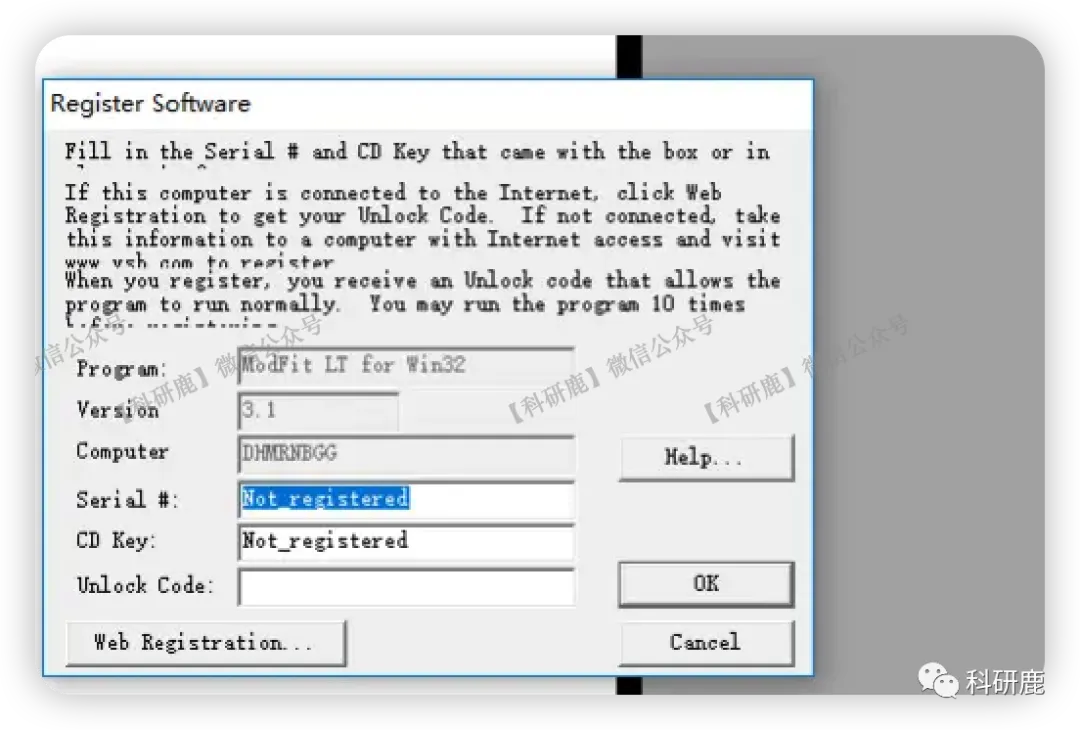
3. 安装成功,自行学习使用即可。
软件下载
请在科研鹿pro公众号后台回复:Modifit

