 夜雨聆风
夜雨聆风
这篇 Science Review 讨论的主题,是人工智能如何进入并改变蛋白质工程。原文的出发点并不是把 AI 描述成一个单独的新工具,而是把它放进蛋白质工程几十年来形成的基本问题中:如何在极其庞大的蛋白质序列和结构空间中,找到具有目标性质的蛋白质。
作者首先回顾了两条前 AI 时代的主线:定向进化和计算蛋白设计。前者依赖突变、筛选和实验测量,能够直接围绕目标性质优化,但吞吐量、时间和成本限制明显;后者依赖物理启发的能量函数与结构建模,搜索范围更广、速度更快,但很难用一个统一能量函数覆盖表达、稳定性、催化活性、动态构象等复杂性质。
整篇综述的核心,是将 AI 蛋白质工程统一解释为两个动作:更高效地搜索蛋白质空间,更准确或更经济地给候选蛋白打分。作者进一步把这一过程写成统计语言:蛋白质设计本质上是从满足目标性质的条件分布中采样。理解了这一点,读者就能把序列生成、骨架生成、反向折叠、全原子建模、零样本评分、表示学习和文库设计放进同一个框架里。
论文基本信息
论文标题:How artificial intelligence is reengineering protein engineering
期刊:Science
发表时间:9 April 2026
DOI:10.1126/science.aec8444
作者:Jennifer Listgarten and Hanlun Jiang
作者单位:Department of Electrical Engineering and Computer Science, UC Berkeley;Center for Computational Biology, University of California Berkeley;UC Berkeley-UCSF Graduate Program in Bioengineering
导读后的核心概括:AI 的角色是搜索和评分
原文开篇先把蛋白质工程的重要性放在生命科学和产业应用的背景下:蛋白质参与能量代谢、基因调控和细胞结构,也可以被改造用于治疗、疫苗、农业、碳固定、生物制造和材料生产。蛋白质工程要完成的任务,是在远短于自然演化的时间尺度内创造或改造具有指定性质的蛋白质。作者强调,这一任务面对的是一个巨大到几乎不可穷举的空间:一个长度只有 100 个氨基酸的小蛋白,也有约 20^100 种可能序列,而其中只有极小一部分能够折叠并表达。
在这个背景下,定向进化和计算蛋白设计分别代表两种互补路径。定向进化用实验测量直接评价目标性质,因此评分与真实设计目标紧密相关,但通常只能围绕已有蛋白做局部探索。计算蛋白设计可以在更广的序列和结构空间中快速搜索,但通常依赖一个近似的物理能量函数,而这个函数未必足以描述催化、动态构象或特定功能。AI 的进入,正是试图同时改善这两个环节:用生成模型或学习到的分布进行更聪明的搜索,用监督模型、生成模型或结构模型对候选变体进行更有效的评分。
因此,本文不是简单罗列 AI 工具,而是先建立一个统一视角:如果目标性质记作 y,蛋白质序列记作 s,那么设计问题可以被看成寻找或采样那些满足目标集合 Y 的序列,也就是从 p(s|y ∈ Y) 中获得样本。后文所有模型路线,都可以被理解为围绕这个条件分布展开。
从传统蛋白质工程到 AI:为什么需要新的搜索方式
原文首先区分了定向进化和计算蛋白设计的基本逻辑。定向进化把自然演化中的突变和选择压缩到实验流程里,通过多轮突变、筛选和测量,把蛋白推向研究者指定的性质。它的优势是不必建立完整的生物物理模型,只要有一个具备相关功能线索的起始蛋白,就可以用实验结果直接指导下一轮优化。
计算蛋白设计则以另一种方式进入问题:它不一定需要一个功能完整的起始蛋白,而是依赖蛋白结构数据库中的统计信息和近似物理模型,在序列和结构空间中进行计算搜索。它通过能量函数给三维构象和序列打分,再选择更有利的候选。作者指出,这种方式在速度和探索范围上不同于定向进化,但也受限于能量函数的表达能力,尤其是当设计目标涉及蛋白动态、量子力学层面的催化效应或复杂功能时,单一评分函数往往过于粗糙。
两条路线的共同难点,是都要在巨大空间中搜索,并对候选变体评分。AI 带来的希望不是绕开这个本质问题,而是改变搜索和评分的方式:一方面,它可以让模型学习怎样从一个序列区域跳到更有希望的区域,而不是只做随机小扰动;另一方面,它可以在实验前预测表达、稳定性、活性等性质,从而让搜索更少依赖盲目筛选。
统计视角:蛋白质设计是从条件分布中采样
进入概念核心后,作者先构造了一个直观场景:假设已有一个体外性质的计算估计 f(s),可以给每条序列 s 预测一个适应度或性质 y。最朴素的做法类似体外的定向进化:从一个序列出发,随机突变,计算 f(s),选择得分更高的变体,再重复。问题在于,这种突变生成方式本身并不知道 f(s) 的形状,因此很难做出跨越式的、协调的变化。
AI 版本的进化算法把这个过程改成了生成模型驱动的搜索。模型不是随机提出突变,而是学习一个序列分布 pϕt(s),并在每一轮根据候选序列的得分更新参数。随着迭代进行,模型逐渐学习哪些序列区域在 f(s) 下表现更好,并学会提出更符合高分区域几何结构的变体。原文特别指出,这里发生了一个语言上的转换:我们不再只是显式搜索某一条序列,而是在搜索一个生成模型的参数,使它能够表示我们想要的序列分布。
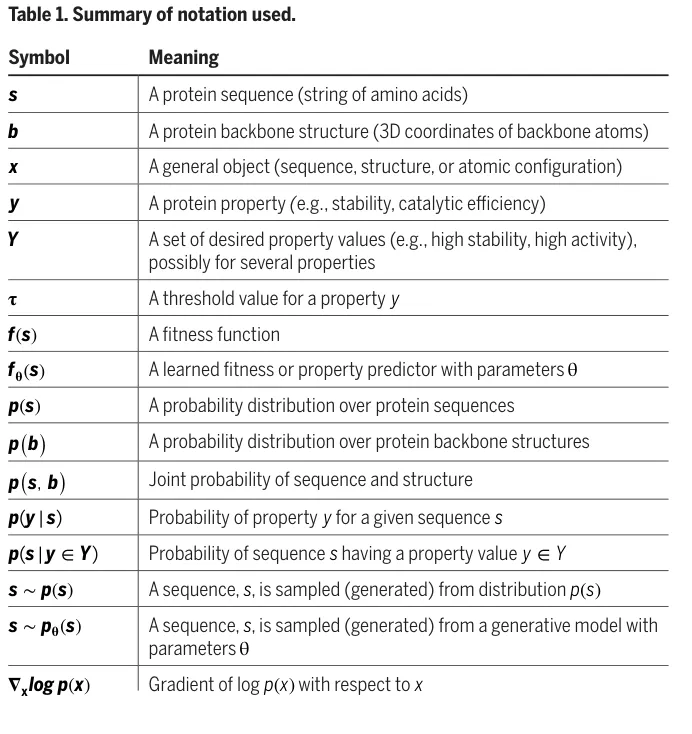
Table 1 的作用,是给后文的统计解释建立共同语言。s 表示蛋白质序列,b 表示骨架结构,y 表示性质,Y 表示满足设计要求的一组性质值,f(s) 是适应度函数,p(s)、p(b)、p(s,b) 则分别表示序列、骨架以及二者联合分布。读者只需抓住一个核心:作者把蛋白质工程中的生成、评分和筛选,都写成概率分布之间的关系。
当性质预测模型具有概率解释时,例如 fθ(s) 可以写成 pθ(y ∈ Y|s),条件自适应采样 CbAS 就可以被解释为一种贝叶斯组合:把一个表示蛋白质先验知识的无条件生成模型 p0(s),与一个表示目标性质的预测模型 pθ(y|s) 结合起来,得到更接近目标的条件分布 p(s|y ∈ Y)。在作者的表述中,AI 蛋白质工程的根本目标,就是估计并采样这一条件分布。
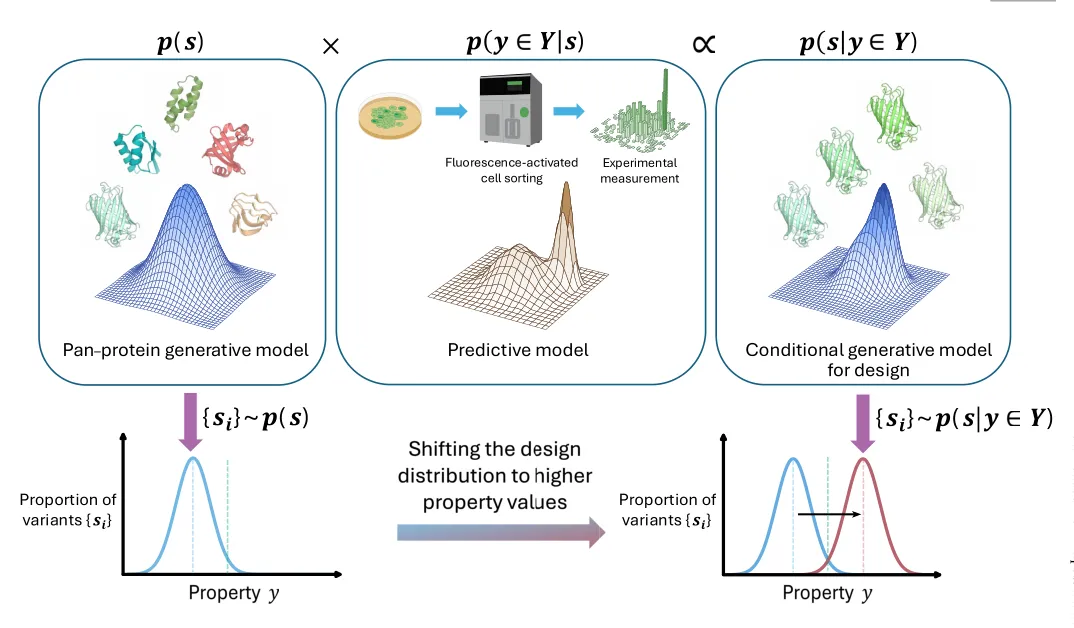
Figure 1 是全文最重要的概念图。左侧的 p(s) 表示一个泛蛋白生成模型,它包含来自大量蛋白序列和结构的背景知识;中间的 p(y ∈ Y|s) 表示性质预测模型,它来自实验测量或监督学习;右侧的 p(s|y ∈ Y) 则表示设计真正想要的分布,也就是更可能满足目标性质的序列。下方的分布曲线说明,AI 设计的目的不是随意生成蛋白,而是把候选变体的性质分布向更高目标值移动。
三种获得条件生成模型的路径
在建立条件分布视角后,原文讨论了三类获得条件生成模型的方法。第一类是在训练时直接把条件变量写入模型,也就是预先决定模型要根据哪些性质生成序列,例如酶分类号、二级结构或配体结合能力。它的优点是概念直接,模型训练后可以直接按条件生成;限制是必须提前知道要纳入哪些条件,并且在训练时就拥有足够的监督数据。
第二类是把已有的无条件模型和性质预测模型组合起来,典型例子是 CbAS。原文把它解释为对贝叶斯规则的使用:用 p(s) 表示泛蛋白背景,用 p(y|s) 表示目标性质预测,再组合出 p(s|y)。这种方式的吸引力在于可以利用已经训练好的大型泛蛋白模型,并在未来根据新的实验数据或目标性质进行定制。不过,贝叶斯规则中的分母涉及对所有可能蛋白序列求和,通常不可直接计算,因此需要变分推断或采样近似。
第三类是作者称为 on-the-fly 的条件化方式:不重新训练一个条件生成模型,而是在采样过程中用性质预测模型提供引导。扩散模型、flow matching、score matching 和随机插值等模型都属于这一讨论范围。它们估计的是对数密度的梯度,而不是直接估计密度本身;在采样过程中,模型把无条件生成分布的梯度与性质预测模型提供的引导项混合起来。原文提醒,这种直觉更自然地适用于连续空间,例如原子三维坐标;对于离散的氨基酸序列,需要采用离散扩散、rate matrix 或连续松弛等专门处理。
作者还特别区分了正式的统计条件化与普通微调。把一个无条件模型在具有某种性质的序列集合上继续训练,有时在实践中有用,但它并不等同于把无条件模型与性质预测分布进行严格的统计组合。这个区别很重要,因为正式的条件化框架能更清楚地说明信息来自哪里、如何组合、什么时候可能失效。
生成模型在蛋白质工程中的具体形态
从家族特异模型到泛蛋白序列模型
原文随后进入工具层面的梳理。最早的一类生成序列方法,是围绕某个野生型蛋白及其同源序列建立多序列比对,再训练 Potts model 等家族特异模型。这类模型能够根据一个蛋白家族的统计规律生成变体,并曾用于 chorismate mutase 等例子。它的局限也很清楚:依赖自然同源序列,依赖一个好的起始蛋白,而且对目标性质的控制主要来自温度等有限参数。
随着 UniRef 等数据库提供大量序列,作者转向泛蛋白模型。MSA Transformer、Neural Potts model 以及后来的蛋白语言模型,试图把跨家族信息放进同一个大型模型中。原文对模型是否是真正意义上的生成模型做了区分:有些 masked language model 虽然被用于生成序列,但在严格统计意义上并不总是可正确采样;相比之下,覆盖完整 masking rate 范围的模型、自回归模型、扩散及相关模型更适合被视为可采样生成模型。ESM3 等多模态模型还同时纳入序列、离散化结构和文本功能注释。
骨架生成模型与反向折叠
在结构设计中,一个典型流程是先从骨架生成模型采样骨架 b,再用反向折叠模型根据骨架生成序列 s。作者用概率链式分解解释这一点:联合分布 p(s,b) 可以分解为 p(s|b)p(b)。RFdiffusion 和 Chroma 等骨架生成模型使蛋白工程社区能够广泛使用以 PDB 为训练基础的骨架扩散模型。它们从噪声中的三维坐标出发,逐步去噪得到结构化骨架,并可以根据对称性、几何约束、结合目标或功能 motif 进行条件生成。
但原文也强调,骨架生成模型的评价仍然困难。常用的 designability 指标会把生成骨架交给反向折叠模型生成序列,再交给结构预测模型判断是否能折回该骨架。这种评价同时受到反向折叠模型和结构预测模型偏差影响,尤其是当研究者希望探索自然蛋白流形之外的区域时,指标未必可靠。
反向折叠模型的任务,是根据给定骨架生成与之兼容的序列。ProteinMPNN 和 ESM-IF1 是原文重点提到的代表,它们使用大规模结构-序列配对数据训练。作者指出,反向折叠的核心难点在于序列与结构之间并非一对一关系,而是多对多关系:一个序列可能具有多个构象状态,一个结构也可能由许多不同序列实现。当前模型通常用单一结构配单一序列训练,这会限制对构象景观和序列多样性的表达。
联合生成与全原子建模
原文进一步讨论了直接估计 p(s,b) 的可能性,也就是同时生成序列和结构。全原子建模试图一次性生成所有原子及其三维位置,这对某些条件很有吸引力,例如活性位点的原子构型、RNA、DNA 或小分子结合目标。作者认为这种方向有技术挑战,例如在序列尚未确定时如何决定要生成多少原子;但它为原子级功能 motif 和非蛋白结合对象的条件化提供了更自然的路径。
生成模型不只用于生成:评分、表示学习与文库设计
原文接着提醒,所谓生成模型并不总是为了直接生成新样本。它们也可以用于评分:如果某条序列在模型看来很像训练数据,那么它可以得到更高的似然或近似似然分数。这类任务被称为 zero-shot prediction,因为没有显式使用目标性质的标注数据。作者指出,在多突变变体的零样本评分基准中,表现较好的模型通常不只是单序列模型,而是纳入结构信息、MSA 信息或两者兼有。
另一类用途是表示学习。研究者可以从生成模型的中间层提取表示,再用于监督学习任务,例如预测结构或蛋白性质。原文同时指出,现有基准数据集存在局限,尤其是许多数据只覆盖接近野生型的少量突变,而 AI 设计真正希望进入的,是离野生型更远、探索价值更高的区域。
在文库设计部分,作者把讨论从模型输出拉回实验现实。即使模型很强,也通常不能只生成一个序列就完成设计;研究者仍需要设计一批候选蛋白,并在实验中筛选。根据问题难度、已有知识和设计区域大小,候选数量可能从几十个到数百万个不等。由此出现两个问题:如何联合设计一批变体,以及如何考虑基因合成成本。
如果设计方法准确率较低,原文指出,有时不如设计一个随机合成过程,而不是精确指定每一条序列。例如通过控制每个位点上 A、C、T、G 的概率,可以在相同合成成本下获得数量级更大的序列集合,但代价是对每条具体序列的控制下降。作者还把这一问题与机器学习引导的定向进化、主动学习和实验预算分配联系起来:核心是怎样在有限时间、劳动力和经费内,选择最值得测量的候选。
什么容易,什么仍然困难:从结合物到酶设计
原文用 What’s easy, what’s hard? 这一节概括了当前进展。蛋白结合物设计是 AI 蛋白质工程中进展较明显的方向之一。AI 之前,计算设计的蛋白结合物文库对治疗相关靶标的命中率通常低于 0.05%;在生成模型和不断扩大的 PDB 支持下,某些案例中的命中率已提高几个数量级,使研究者可以在微孔板层面做常规表征,而不是完全依赖劳动密集型高通量筛选。
但作者马上补充,生成模型本身并不足够。当前流程仍然高度依赖后处理筛选,包括调用 AI 结构预测器以及传统生物物理标准。将蛋白设计为结合任何类型生物分子也仍然更难,尤其是 DNA、RNA 和小分子,因为相关复合物结构数据不足。成功设计出的结合物也多是小型、球状、以螺旋和片层为主的蛋白,而天然蛋白如抗体常常依赖 loop 进行分子识别;通用模型还不能稳健设计柔性 loop 和内在无序区域。
酶设计被作者描述为更困难的问题,因为它要求非常精确的原子级知识,尤其是活性位点构型。对于简单且研究充分的反应,化学专家可以通过量子力学计算构造理想化活性位点,也就是 theozyme;但对于复杂反应,常见策略只能从已知酶中提取活性位点,再以此条件化生成完整酶。这样的策略可以用于小型化、提高稳定性或单体化,但并不足以为未知反应设计新的催化活性,而且往往仍需要定向进化进一步优化。
原文还提到另一种基于功能注释的酶设计策略,即不显式建模活性位点,而是根据 Enzyme Commission number 等功能标签直接生成序列。作者对这一策略的限定也很清楚:它不太可能为此前未知的反应生成真正有功能的酶,因为模型仍然依赖已有功能类别和训练数据。
展望与讨论:数据、泛化和评估仍是瓶颈
在 Outlook and discussion 中,作者把当前 AI 蛋白质工程的依赖关系讲得很直接:许多设计流程依赖结构预测模型,要么在生成循环中显式调用,要么在事后筛选中使用。然而结构预测模型扎根于自然蛋白宇宙,未必足以广泛判断人工设计序列是否有用。结构预测领域的进展,包括与冷冻电镜数据或分子动力学数据结合的方法,可能会继续影响蛋白质工程,但这些进展也提醒我们,AI 离不开昂贵、宝贵且最好公开的实验数据库。
作者反复强调泛化问题:AI 模型能否走向蛋白空间中“新的”区域,本身就是难以定义和验证的问题。由于训练数据附近往往表现更好,而远离训练分布时可靠性下降,原文提出可以理性融合生物物理模型与 AI 模型。前者理论上应更均匀地适用于蛋白空间,后者则在接近训练数据的区域表现突出;二者结合可能比单独依赖任一方更合适。
评估也是原文最后反复指出的问题。生成模型本就难以评价,而蛋白设计的真实评价常常需要湿实验,成本高、周期长,因此多数论文难以系统、现实地比较不同方法。作者提到,结构预测领域的 CASP 竞赛之所以推动巨大,是因为可以在不合成蛋白的情况下做评估;但设计任务往往没有这样便宜且可靠的评估标准。作者因此呼吁更强的体外基准问题、更有用的指标,以及包含湿实验验证的竞赛,同时也鼓励新方法论文提供更多 baseline。
结尾总结:这篇综述真正想建立的框架
从全文看,这篇综述最重要的贡献不是给出某一个模型排行榜,而是把 AI 蛋白质工程重新组织成一个统计问题。设计蛋白,就是从满足目标性质的条件分布中采样;生成模型提供候选空间,性质预测模型提供目标方向,贝叶斯组合或采样引导则把二者连接起来。
第二个重要信息是,序列生成、骨架生成、反向折叠、全原子建模、零样本评分、表示学习和文库设计并不是彼此孤立的工具箱。它们都围绕搜索、评分和条件化展开,只是处理的对象不同:有的处理序列,有的处理骨架,有的处理结构-序列联合分布,有的处理实验预算和合成成本。
第三个结论是,AI 已经显著改变某些蛋白质工程任务,尤其是结合物设计和候选筛选效率;但在酶设计、柔性 loop、内在无序区域、非蛋白结合目标、远离训练分布的泛化以及真实评价指标上,原文仍然保持谨慎。作者的态度并不是简单宣称 AI 已经解决蛋白质工程,而是强调 AI 正在把这个领域推进到一个更明确的问题表述中:如何利用有限数据、有限实验和有限计算,更有效地逼近我们真正想要的条件分布。
