 夜雨聆风
夜雨聆风
对接给出结合假设,MD检验动态稳定性。关键残基不一致,不一定是结果错了。
做分子对接和分子动力学模拟时,很多人都会遇到一个问题:分子对接里看到的关键残基,和分子动力学分析出来的关键残基不完全一样。
比如,对接结果显示小分子和 A、B、C 几个残基形成了氢键或疏水作用;但做完分子动力学模拟后,氢键占有率、接触频率或者 MM/GBSA 残基能量分解结果却提示 D、E、F 这些残基贡献更明显。这时候很多人会疑惑:是不是对接结果错了?是不是分子动力学结果不可靠?正常情况下,两者是不是应该完全一致?
其实,这个问题非常常见。它不一定说明计算错了,而是要看对接和MD分别回答的是什么问题。
一、对接看的是“可能怎么结合”
分子对接更像是一个初始预测工具。它通常基于一个相对固定的蛋白结构,把小分子、肽段、多糖或另一个蛋白放到可能的结合区域中,预测一个评分较好的结合姿势。
所以,对接图里看到的关键残基,更多代表的是:在这个初始结合构象中,哪些残基离配体比较近,可能形成氢键、疏水作用、静电作用或 π-π 相互作用。也就是说,对接结果可以帮助我们判断配体可能进入哪个口袋、初始可能接触哪些残基、结合姿势大概是什么样。
但它毕竟是一个相对静态的结果。对接图上出现一条氢键,并不代表这条氢键在真实水环境和动态波动中一定长期稳定存在。因此,对接结果更适合作为初始结合假设和候选相互作用残基。
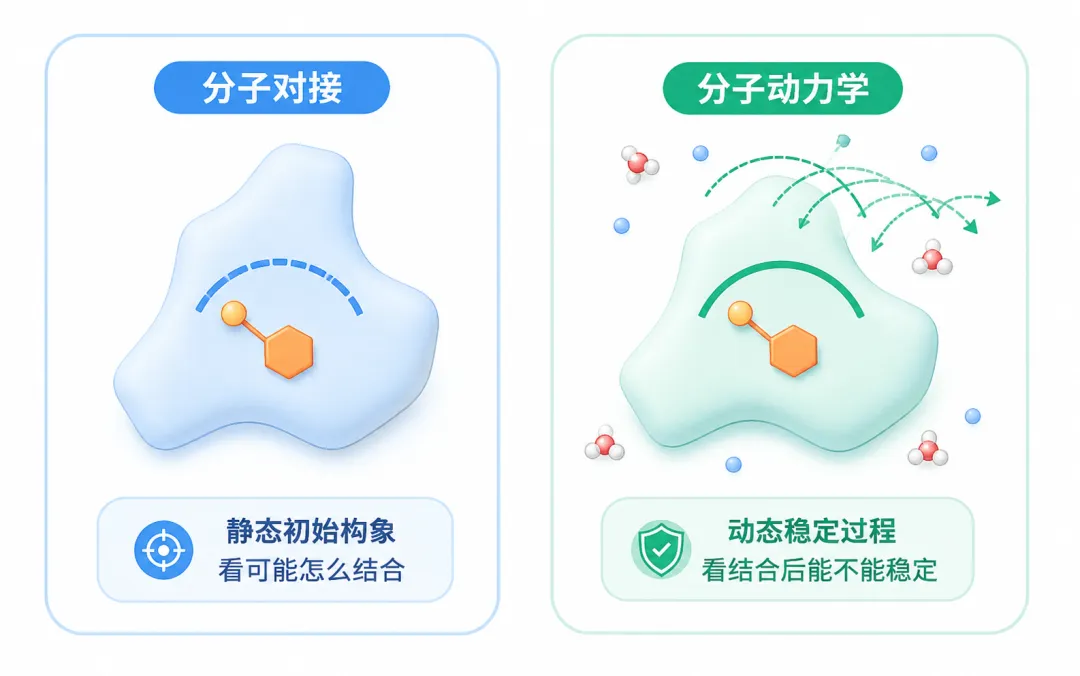
图1 分子对接更偏向于预测初始结合姿势,分子动力学模拟则进一步观察该结合模式在动态环境中的稳定性。
二、MD看的是“结合以后稳不稳定”
分子动力学模拟关注的是另一个问题:这个结合模式在水环境、离子环境、温度和压力条件下,能不能稳定存在。
在MD过程中,蛋白不是静止的,配体也不是固定的。蛋白侧链会摆动,结合口袋会发生局部开合,配体构象会调整,水分子和离子也可能参与竞争作用。所以,模拟跑起来之后,初始对接构象可能会发生变化。
有些对接图上看起来很明显的氢键,可能跑一段时间后就断开了;有些对接时不太显眼的残基,反而在构象调整后形成了更稳定的接触;还有一些残基虽然没有漂亮的氢键,但通过疏水作用、范德华作用或静电作用,对结合稳定性贡献很大。
因此,MD分析出来的关键残基,通常更偏向于动态过程中真正稳定参与相互作用的残基。
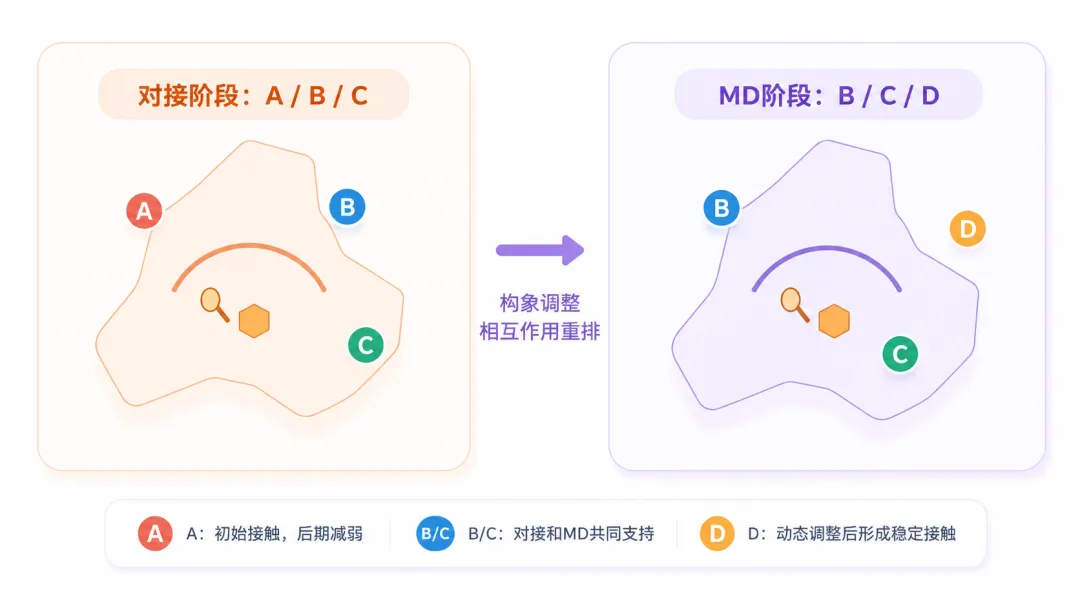
图2 对接和MD得到的关键残基不完全一致并不罕见。部分初始相互作用可能在模拟过程中减弱,也可能出现新的稳定接触残基。
三、很多课题是先做对接,再用MD进一步验证
在实际课题中,很多人并不是一开始就把分子对接和分子动力学全部做完,而是一步一步推进的。
更常见的流程是:先做分子对接,看看配体是否能够进入目标口袋,结合姿势是否合理,周围是否存在氢键、疏水作用等相互作用。如果对接结果看起来比较有希望,比如结合区域合理、打分较好、相互作用残基也有一定基础,研究者才会更有信心进一步做分子动力学模拟。
所以,很多时候并不是“对接和MD一开始就应该完全一致”,而是先用对接提出一个可能的结合假设,再用MD去检验这个假设在动态环境中是否稳定。
也正因为如此,后续出现关键残基不完全一致,其实并不奇怪。对接阶段看到的是初始构象中的候选残基,而MD阶段看到的是体系经过构象调整后,哪些残基真正能够长期维持结合。
这里要分清两种情况:如果项目目前只做到了对接阶段,那当然可以先基于对接构象分析候选相互作用残基;但如果后续已经做了MD,尤其是对接构象经过MD后形成了较稳定的代表性构象,那么关键残基的讨论就不能只停留在最初的对接图上。
更常规的做法是:用对接结果说明“为什么选择这个结合模式进入MD”,再用MD后的稳定构象、轨迹分析和能量分解来判断“哪些残基更可能真正维持结合”。
实际写文章或做报告时,如果一个体系既做了分子对接,也做了分子动力学模拟,那么在判断关键残基时,通常应更侧重分子动力学结果。因为对接看到的是静态初始结合姿势,而MD看到的是这个结合模式在水环境、蛋白柔性和构象波动中能不能稳定下来。
比如,对接图里某个残基形成了氢键,但在MD过程中氢键占有率很低,接触时间很短,距离也很快拉开,那么这个残基就不适合被直接写成“核心关键残基”。
相反,如果某个残基在对接图中不算特别显眼,但在MD过程中长期保持接触,或者在残基能量分解中贡献明显,那么它反而更值得重点讨论。
因此,如果同时有对接和MD结果,一般不能只拿对接图来定关键残基,而应以MD中的动态稳定性证据为主,同时结合对接构象解释初始识别过程。
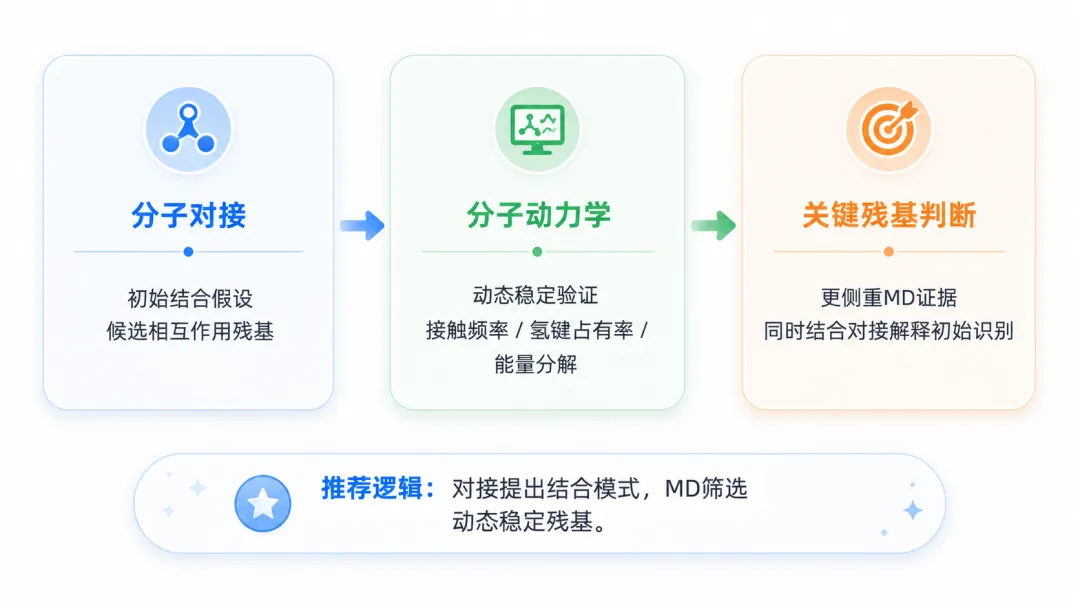
图3 很多课题会先用对接提出候选结合模式,再用MD验证动态稳定性;如果MD已经完成,关键残基判断通常更侧重MD稳定构象和轨迹证据。
四、但这不代表对接结果可以完全忽略
需要注意的是,更侧重MD结果,并不是说对接结果没有价值。
对接仍然有它的作用。它可以告诉我们:配体最初可能进入哪个结合区域,可能采取什么结合姿势,周围有哪些候选相互作用残基。
MD则进一步判断这些初始相互作用是否能在动态过程中维持。
所以,两者不是互相否定,而是前后递进:对接负责提出问题,MD负责进一步验证;对接给出初始构象,MD检验动态稳定性。
如果对接和MD在结合口袋、主要作用类型和部分核心残基上具有一定连续性,说明这个结合模式相对合理。
如果两者完全对不上,比如对接显示配体在A口袋,MD后配体跑到B口袋;或者对接残基和MD残基在空间上完全不相关,那就需要重新检查模型是否合理。
可能的问题包括:对接口袋设置不合适、蛋白前处理不充分、配体质子化或电荷参数不合理、力场参数有问题、MD模拟时间太短,或者体系没有充分平衡。
五、正常情况下,两者应该“大方向一致”
理想情况下,分子对接和分子动力学结果应该具有一定一致性。
但这里说的一致,不是指每一个残基名字都完全一样,而是指:结合区域大体一致;主要作用模式大体一致;部分核心残基能够在MD中得到保留或验证;MD中新出现的残基与原结合口袋存在空间连续性。
如果只是具体残基名单有所变化,但配体仍然稳定停留在原口袋,整体结合模式没有明显跑偏,这通常是可以接受的。
因为MD过程中蛋白侧链会动,配体也会调整。相互作用残基发生一定变化,反而是动态模拟中很常见的现象。
真正需要警惕的是“大方向完全不一致”。比如配体在MD中明显漂出口袋;对接中的主要相互作用很快全部消失;残基能量分解贡献最大的位点和对接口袋完全不在一个区域;配体结合姿势发生明显翻转,且无法形成稳定接触网络。
这种情况就不能简单说是“动态调整”,而要考虑初始模型或模拟设置是否存在问题。
六、不同软件分析出来的残基为什么也会有差异?
还有一种常见情况是:同一个体系,用不同软件或不同分析方法,得到的关键残基也不完全一样。
这也不奇怪。因为不同软件的对接算法、打分函数、口袋设置方式、相互作用识别标准都可能不同。即使是同一条MD轨迹,不同分析方法使用的距离阈值、角度标准、氢键判定规则、接触定义和能量分解方法也可能不同。
比如,一个软件可能按距离判断接触,另一个软件可能同时考虑角度;一个方法强调氢键,另一个方法更容易突出疏水或范德华贡献;MM/GBSA和MM/PBSA 的计算模型不同,残基分解结果也可能有一定差异。
所以,不同软件给出的残基结果大体逻辑可以类似,但不一定逐个残基完全一致。
实际分析时,不能只机械比较残基名字,而要看这些残基是否位于同一结合区域,是否参与同一类相互作用网络,是否在MD过程中具有稳定接触,是否在能量分解中有合理贡献,是否与已有实验或文献背景相符。
判断关键残基,本质上不是看软件自动输出了哪些名字,而是看这些残基是否有足够证据支持它们参与结合稳定。
七、遇到不一致时,可以把残基分成三类
实际写文章时,最稳妥的方法不是强行让对接和MD完全一致,而是把残基分层解释。
第一类是对接和MD都支持的残基。这类残基可信度最高。如果一个残基在对接中形成明显相互作用,在MD中也有较高氢键占有率、接触频率或能量贡献,那么它可以作为重点讨论的核心残基。
第二类是只在对接中出现的残基。这类残基不一定没有意义,但要谨慎解释。它们可能参与了初始识别,也可能只是静态构象中的瞬时接触。如果在MD过程中接触频率低、距离很快拉开,就不适合直接说成决定性关键残基。
第三类是只在MD中突出的残基。这类残基也很重要。它们可能是在体系动态调整后形成的稳定接触,也可能通过疏水、范德华或静电作用维持结合稳定性。尤其是能量分解中贡献明显、空间位置又合理的残基,可以作为动态稳定残基进行讨论。
这样处理以后,结果就不会显得“对接和MD互相矛盾”,而是变成一个更合理的逻辑:对接提示潜在结合模式,MD筛选动态稳定相互作用。

图4 对接和MD残基不完全一致时,可以按证据来源进行分层解释,而不是简单判断某一个结果错误。
八、报告里可以怎么写?
如果对接和MD结果大方向一致,但具体残基不完全相同,可以这样写:
分子对接结果显示,配体能够进入目标蛋白的结合口袋,并与周围多个残基形成氢键、疏水作用或静电相互作用,提示该区域可能为潜在结合位点。进一步的分子动力学模拟表明,配体在模拟过程中整体保持在该结合区域内,说明初始对接构象具有一定稳定性。与此同时,随着蛋白侧链和配体构象的动态调整,部分初始相互作用发生减弱,而另一些残基在模拟过程中表现出更高的接触稳定性或能量贡献。因此,在关键残基判断上,应以MD过程中的动态稳定性证据为主要依据,同时结合对接构象解释初始识别过程。
如果两者差异比较大,可以这样写:
分子对接结果提示配体可能结合于目标口袋,但后续分子动力学模拟显示,初始相互作用在模拟过程中稳定性不足,部分对接残基的接触频率较低,配体结合姿势也发生了一定调整。这提示初始对接构象可能只代表一种潜在结合假设,仍需结合轨迹稳定性、结合自由能、残基能量分解及实验信息进一步判断其合理性。
总结
分子对接和分子动力学得到的关键残基不完全一致,是很常见的现象。
正常情况下,两者应该在结合区域、主要作用模式和部分核心残基上保持一定一致性,但不需要每一个残基完全一样。
很多课题确实是先做对接,看结合模式有希望后,再进一步做MD。这个流程本身没有问题。关键是:如果后续已经做了MD,那么关键残基分析就不应该只停留在最初的对接图上,而应更多回到MD稳定构象和轨迹证据。
但对接结果也不能完全丢掉。它是初始结合模式的重要参考,可以帮助解释配体最初如何进入口袋、可能与哪些残基发生识别。
总结:对接给出结合假设,MD检验动态稳定性。关键残基不一致,不一定是结果错了,关键要看这个结合模式在动态过程中是否站得住。
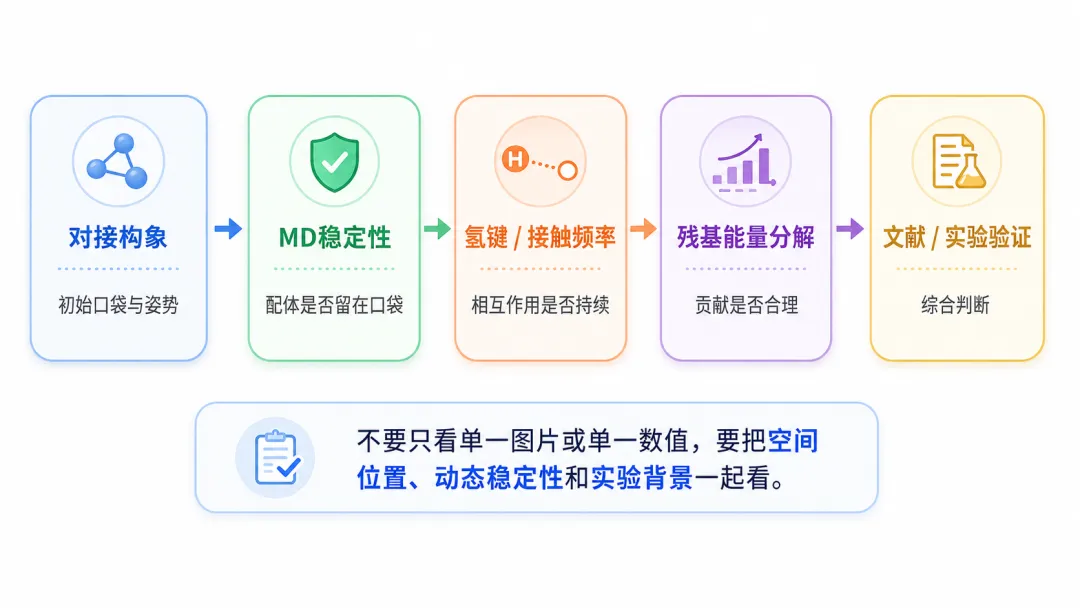
图5 判断关键残基不能只看单一图片或单一能量值,应结合空间位置、动态稳定性、能量贡献和实验背景综合分析。
参考资料
[1] Ferreira LG, dos Santos RN, Oliva G, Andricopulo AD. Molecular Docking and Structure-Based Drug Design Strategies. Molecules. 2015.
[2] Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery. 2015.
[3] Wang E, Sun H, Wang J, et al. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chemical Reviews. 2019.
本文中配图为辅助理解绘制的示意图,不代表某一具体体系的真实计算结果。实际项目中,关键残基判断应结合蛋白结构、配体性质、模拟参数、轨迹稳定性、能量分解结果及实验背景综合分析。如有疏漏欢迎指正交流。

