 夜雨聆风
夜雨聆风
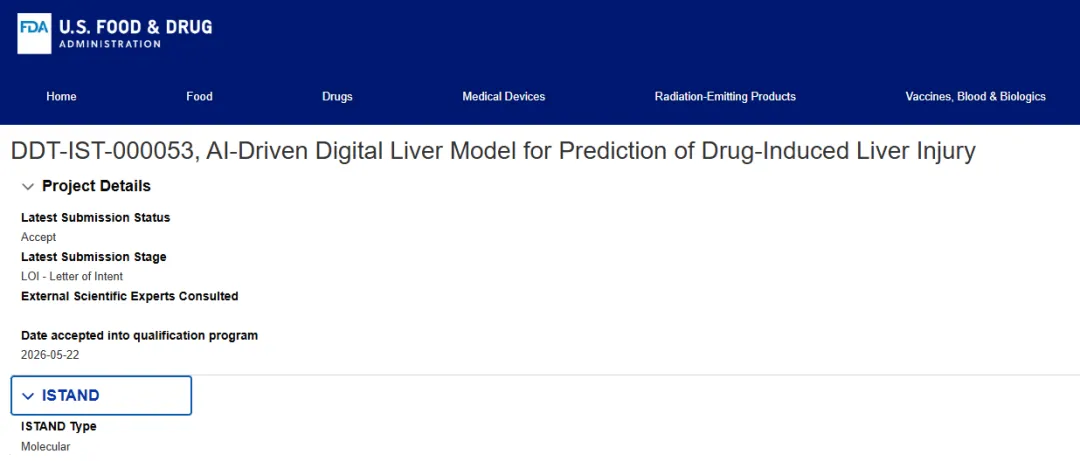
2026 年 6 月 3 日,FDA 药品评价与研究中心(CDER)对外宣布,首次受理一项 in silico(计算机模拟)药物研发工具(Drug Development Tool, DDT)进入 ISTAND 资格认定项目。这项工具是一个 AI 驱动的数字肝脏模型,用于预测药物性肝损伤(Drug-Induced Liver Injury, DILI)。这是 AI 安全预测工具正式进入 FDA 的监管级资格认定通道,对所有关注上市后监测与获益风险评估的药物警戒从业者,都是一个值得记下的时间节点。
更值得细读的,是 FDA 同步公开的那封 LOI 裁定函(编号 DDT-IST-000053-LOI-2,2026 年 5 月 22 日签发)。新闻稿讲的是"发生了什么",裁定函讲的是"FDA 到底怎么审 AI 模型"。后者才是 PV 与监管科学从业者真正能学到东西的地方。下面把这件事拆开讲清楚。
FDA 这次做了什么
CDER 受理了一封意向书(Letter of Intent, LOI),让这项 AI 数字肝脏模型正式进入 ISTAND 项目的 DDT 资格认定流程。申请方是位于美国马萨诸塞州 Somerville 的 Absentia Labs, Inc.,递交人为 Robert Betancort。整个流程依据《联邦食品药品和化妆品法案》(FD&C Act)第 507 条进行,这一条款由 2016 年《21 世纪治愈法案》新增,是 FDA 当前运行 DDT 项目的法律基础。
ISTAND 全称是 Innovative Science and Technology Approaches for New Drugs,即新药创新科学与技术方法项目。它的定位很特殊:专门接纳那些无法套进现有评估和申报路径的新型 DDT,比如不属于传统生物标志物、也不属于临床结局评估的工具。换句话说,ISTAND 是 FDA 为"还没有现成监管框架的新工具"留的一扇门。
这次进来的工具被定义为一种新方法学(New Approach Methodology, NAM)。它和 FDA 长期推动的动物试验 3R 原则一致,即替代(replacement)、减少(reduction)、优化(refinement)。需要强调的是,受理 LOI 只是认可它有资格进入流程,并不等于这个工具已经被批准或被认定可用。这是起点,不是终点。
值得 PV 人注意的一个细节:FD&C 第 507 条带有透明度条款,申请方递交的部分信息会被依法公开在互联网上。这正是我们今天能读到这封裁定函全文的原因,也意味着 ISTAND 通道里的 DDT 进展是可以持续公开跟踪的。
把模型看清楚:它到底预测什么
新闻稿笼统地说这个工具"比对化学结构",但裁定函给出了精确得多的使用情境(Context of Use, COU)。FDA 在信中甚至直接给申请方推荐了一版规范的 COU,核心要点如下:
这是一个用于评估成人小分子候选药物诱发肝损伤风险的工具,产出可纳入候选药物的新药临床试验申请(IND)。模型给出的是分类预测,把候选化合物划入高、中、低三个 DILI 风险等级,依据是化合物层面的理化性质、分子结构和临床毒理数据。它有一个明确的适用前提:当候选化合物与既往已表现出临床肝毒性的某个药物属于同一药理学类别时使用。其分类结果作为补充证据,支持对临床前安全性研究的解读。
这里有几个关键信息:第一,它的输出不是黑白二分,而是高/中/低三级分类;第二,它有清晰的适用边界(同药理学类别 + 已知肝毒性参照物 + 成人 + 小分子);第三,它的定位始终是"补充证据",服务于 IND 阶段的研发决策,而不是替代既有毒理研究。
从裁定函附录还能看出,模型整合了理化性质、分子指纹(molecular fingerprint)和临床前毒理数据三类输入,并能给出可解释性结果(explainability),说明各特征对整体毒性画像的贡献。这已经不是一个简单的结构相似度工具,而是一个带置信度评分和特征归因的预测系统。
时间线:一封裁定函背后的九个月
裁定函披露的时间线,清楚展示了一个 DDT 进入 ISTAND轨迹:
2025 年 11 月 18 日,Absentia Labs 递交 LOI-2。
2025 年 12 月 15 日,FDA 出具可审性状态备忘录(Reviewability Status Memo),判定为"可审"。
2026 年 2 月 13 日,FDA 发出信息征询函(IR letter),就模型与对照工具的比较等问题提问。
2026 年 5 月 22 日,FDA 签发 LOI 裁定函,正式受理进入资格认定项目。
2026 年 6 月 3 日,CDER 对外发布新闻。
从递交到受理走了约半年,且中间经历了一轮正式的信息征询。监管对 AI 工具的态度是开放但审慎的,每一步都留痕、都有书面裁定。
真正的干货:FDA 审一个 AI 模型,提了 16 条意见
裁定函附录里,FDA 列出了 16 条考虑与建议,要求申请方在下一步的资格认定计划(Qualification Plan, QP)中逐条回应。这 16 条,几乎就是一份"监管如何评估 AI 安全模型"的清单,对想把 AI 用进合规场景的 PV 人可以参考一下。把它们归纳为五个维度:
使用情境要精准
COU 应当是一句简短的"工具该如何用于药物研发"的陈述,不应混入工具描述和使用示例。申请方原来的 COU 写得太杂,FDA 直接帮它改写了一版。监管对 AI 工具,第一关就是逼你把"到底用在哪、能支撑什么结论"讲清楚。
分析验证要拿数据说话
FDA 要求提供模型的分析验证数据,覆盖准确性、预测值、可重复性(repeatability)和可再现性(reproducibility)等性能指标。还追问:置信度评分(confidence score)是怎么算的,它反映的是模型预测本身,还是反映训练所用历史数据的数量与质量,并建议设定一个置信度阈值,低于该阈值的结果视为无效。
模型必须锁定
用于 FDA 资格认定的最终模型版本必须被锁定(locked down),且用于开发最终模型的数据(药物及其结构、DILI 标签)不能再被复用于资格认定。这等于在说,监管认可的不是一个会持续变动的模型,而是一个版本冻结、可被独立验证的模型。
统计严谨性不能少
FDA 要求补充化合物排除原因的数量与占比,解释"DILI 分类模糊或矛盾"这一排除场景,讨论排除是否引入系统性偏倚、各药理学类别是否被充分代表、对模型泛化能力有何影响;还要求按药物类别做亚组分析,并在统计分析计划(SAP)中详述 AI 模型的输入输出、方法框架、调参策略与全部性能指标定义。
机制与可解释性要落地
FDA 希望模型不仅指出毒性特征贡献,还能基于历史数据提示可能发生的肝毒性类型,以利于后续机制研究;并追问模型是否覆盖所有类型的 DILI,包括免疫介导型 DILI(immune-mediated DILI);还要求说明当模型结果与其他临床前数据相矛盾时,使用者该如何解读。
对 PV人的影响
这条新闻表面上是临床前研发的事,但药物警戒人没有理由忽略它,原因有三。
肝毒性是贯穿全生命周期的核心安全信号。DILI 不只是临床前问题,它也是上市后信号检测、获益风险评估、定期安全性报告中反复出现的重点。一个能在早期更准确分级 DILI 风险的工具,会改变某些药物进入临床和上市的安全底色,进而影响后续的安全性监测预期。
AI 正在从"提效工具"变成"监管评估对象"。过去 PV 圈讨论 AI,多停留在病例处理自动化、文献监测、智能数据提取这类内部提效层面。而这次,AI 以一个待认定 DDT 的身份,进入 FDA 的正式资格认定流程,并被要求做模型锁定、分析验证、偏倚分析、亚组分析。这套要求,恰恰是未来任何 AI 工具想进入 PV 合规场景都绕不开的关卡。读懂 FDA 这 16 条,就是提前看懂了 AI 在药物安全领域的合规标尺。
证据权重思维是 PV 的基本功,也适用于看待 AI 工具。FDA 反复强调这个模型提供的是"补充证据",不是单一决定因素,还专门追问"结果与其他数据矛盾时怎么办"。这恰恰是成熟 PV 人评估任何安全性证据时的标准姿态:任何单一来源,无论是 AI 还是传统模型,都只是证据链的一环。
结语
FDA 首次让一个 AI 数字肝脏模型走进 ISTAND 资格认定流程,并用一封 16 条意见的裁定函,第一次完整展示了监管如何评估一个 AI 安全预测模型:要锁定版本、要分析验证、要查偏倚、要讲清置信度和可解释性。
读懂它,提前拿到了 AI 进入药物安全合规场景的标尺。
原文链接:https://force-dsc.my.site.com/ddt/s/ddt-project?ddtprojectid=247
