 夜雨聆风
夜雨聆风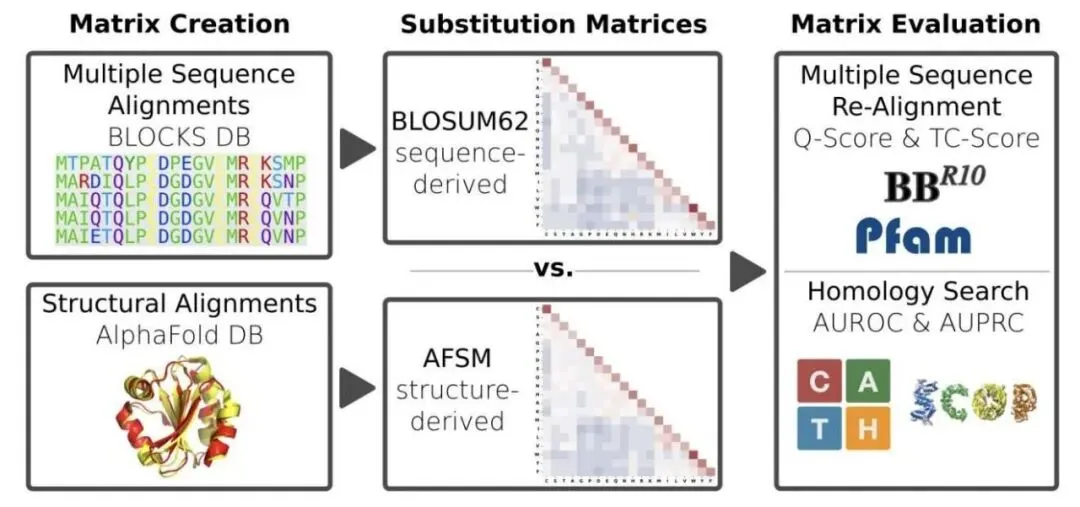
<<< 左右滑动见更多 >>>
榴莲忘返AIDD
供稿 | 柠檬青年
审稿 | 吉星
目录
用海量 AlphaFold 结构数据构建的氨基酸替换矩阵 (AFSM),其性能与 30 年前的 BLOSUM62 几乎没有差别,这说明在今天,死磕替换矩阵可能已经意义不大了。 AlphaUnfold 用短时高压分子动力学模拟,为 AlphaFold3 静态结构提供了快速、有效的物理稳定性「压力测试」,能快速识别出结构中的脆弱区域。 这是一种全新的抗体性质预测框架,它在推理时通过上下文学习动态适应新数据,巧妙地解决了实验中的批次效应问题,并且模型又小又快。 AblateCell 是一个能自动复现生物学 AI 模型并精准找出其核心有效模块的智能体,解决了代码验证中耗时耗力的难题。 RosettaSearch 把大语言模型变成了化学家,让它根据结构预测软件的报错反馈反复修改蛋白序列,无需微调就将设计成功率提升了 2.5 倍。
1. AlphaFold 建新矩阵,效果竟和 30 年前一样?
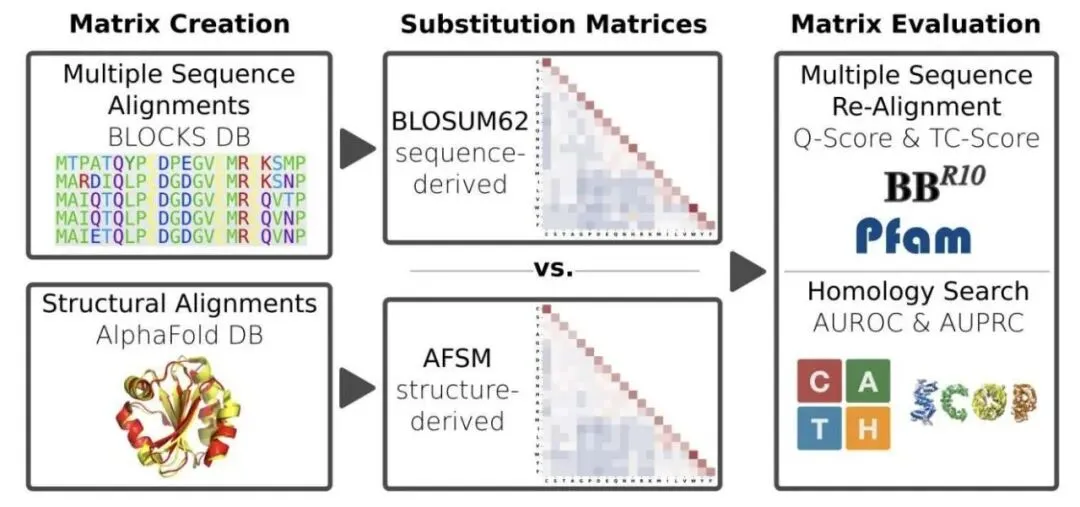
做生物信息学和计算生物学的,工具箱里总有几件用了几十年的「老古董」,氨基酸替换矩阵就是其中之一,比如大名鼎鼎的 BLOSUM62。它告诉我们,在进化过程中,一个氨基酸有多大概率被另一个替换。最近,有研究者想,既然我们现在有了 AlphaFold 预测的海量、高质量的蛋白质三维结构,能不能用这些结构信息,做一个比基于序列的 BLOSUM 更牛的替换矩阵?
想法很直接。他们拿了超过 20,000 个蛋白,分属近 300 个家族,用它们的预测结构进行了约 66 万次结构比对,最终得到了包含近 7000 万个氨基酸配对信息的数据集。这个工作量是巨大的。基于这些数据,他们构建了一个新的矩阵,叫做 AFSM (AlphaFold Substitution Matrix)。
但结果出来,所有人都愣了一下。这个用最新技术、海量数据做出来的 AFSM 矩阵,和 1992 年的 BLOSUM62 简直像一个模子刻出来的。两者相关性高达 96%,矩阵里 48% 的数值完全相同,绝大多数差异也仅仅是 ±1。
当然,光看矩阵数值不能说明全部问题,得拉出来实战检验。研究者在五个基准测试中比较了 AFSM 和 BLOSUM62 的性能,涵盖了多序列比对(MSA)重建和远程同源搜索这两个经典任务。结果是,两者的表现几乎一模一样。比如在 BAliBASE 基准测试中,Q-score 分别是 71% 和 70%;在最难的 CATH S20 同源搜索中,ROC AUC 都是 64%。没有任何显著差异。
更有意思的是研究者做的一个「对照组」实验。他们设计了一个极简的「单位矩阵」:氨基酸匹配就得 5 分,不匹配就得 0 分或 -1 分。这种简单粗暴的规则,在构建多序列比对时,效果居然不比精雕细琢的 BLOSUM62 差。
这说明了什么?它告诉我们,在很多情况下,比对算法的性能主要来自一个笼统的「保守性信号」(匹配得分远高于错配)以及合理的空位罚分(gap penalty),而不是替换矩阵里那些精细的、不同氨基酸之间的替换偏好。
这就引出了一个核心观点:替换矩阵的价值,取决于你手头有多少数据。当你只有一个蛋白序列,想在茫茫数据库里找亲戚时(序列数据稀疏),一个好的替换矩阵能提供宝贵的先验知识。但如果你已经有了一个包含几十上百条序列的 MSA(数据充足),序列本身的变化模式已经提供了足够丰富的信息,替换矩阵那点先验知识就显得无足轻重,甚至可能带来噪音。
那么,在处理真正棘手的远程同源搜索问题时,谁才是王者?答案是蛋白质嵌入模型,比如 ProtTrans T5。这些大语言模型将每个蛋白质序列转化为一个高维向量(embedding),捕捉了更深层次的进化和结构信息。在最难的 CATH S20 测试集上,基于嵌入的搜索方法 ROC AUC 达到了 90%,而 AFSM 和 BLOSUM62 只有 64% 左右。这个差距是压倒性的。
所以,这篇论文的标题《无事生非》(Much ado about nothing)起得非常贴切。它不是在否定 BLOSUM62 的历史贡献,而是在用无可辩驳的数据告诉我们:在 2024 年,试图通过更大、更新的数据去优化一个诞生于「小数据」时代的工具,其边际效益几乎为零。工具的演进,终究要跟上时代的步伐。对于挑战性的生物学问题,我们的目光应该从替换矩阵,转向像蛋白质嵌入这样更强大的新范式了。
📜Title: Much ado about nothing: Modeling amino acid replacement with predicted protein structures
🌐Paper: https://doi.org/10.1093/bioinformatics/btag188
2. 给 AlphaFold3 模型做个「压力测试」,5 纳秒见分晓
我们每天都在用 AlphaFold3 (AF3) 生成海量的蛋白质结构。这些模型看起来很漂亮,但作为一个在一线做研发的科学家,我脑子里总会冒出一个问题:这些静态的 3D 图片在真实的、动态的细胞环境里站得住脚吗?一个预测出来的结构,和它真正能稳定存在,是两码事。
这篇 AlphaUnfold 的论文,就试图回答这个问题。他们提出的方法很直接,甚至有点「暴力」:给 AF3 预测的蛋白模型施加巨大的物理压力,看它会不会散架。
这套方法的原理很简单,就像是给蛋白质做「碰撞测试」。
汽车工程师设计了一款新车,不会只看设计图,而是会把样车拿去狠狠地撞一下,看看哪里最先变形,哪里的结构最脆弱。AlphaUnfold 做的就是类似的事情。它把 AF3 给出的蛋白结构模型,丢进一个充满水分子的虚拟环境里,然后把压力瞬间加到 1000 个大气压。这个压力非常大,相当于深海一万米的压强。
在这么大的压力下,蛋白质结构中任何不稳定的部分都会被迅速放大。整个过程只需要模拟 5 纳秒(nanosecond),这在分子动力学(Molecular Dynamics, MD)模拟里算是「闪电战」了。然后,系统会自动计算并报告,这个蛋白结构变形了多少。
核心发现:AF3 的自信程度,和蛋白的抗压能力直接挂钩。
研究者发现了一个清晰的规律:AF3 给出的 pLDDT 分数(可以理解为 AI 对其预测结果的自信程度,分数越高越自信)和蛋白质在模拟中的结构变化(用 RMSD,即均方根偏差来衡量)之间,存在一个非常漂亮的反比关系。
他们甚至给出了一个公式:RMSD = 35 − 0.34·pLDDT。简单来说,pLDDT 分数每高 10 分,结构在压力下的变形程度就减少约 3.4 埃。这个结果告诉我们,AF3 的自信是有物理依据的,高 pLDDT 的区域通常也更稳定。
为什么说它是个「快捷方式」?
做过 MD 模拟的人都知道,这东西非常耗费计算资源。为了得到可靠的结果,一个常规模拟跑上几百纳秒甚至微秒是家常便饭。谁有时间给成百上千个候选设计都跑一遍长时程模拟?
AlphaUnfold 的聪明之处在于,它证明了 5 纳秒的高压模拟,虽然粗暴,但识别出的「结构软肋」和花几十倍时间跑的 200 纳秒常规模拟结果高度一致。它能快速告诉你蛋白质的哪个 loop 不稳定,哪个 domain 容易摇摆。这对于高通量的筛选来说,价值巨大。
从「看整体」到「抓痛点」,这才是对我们最有用的地方。
除了给出一个总体的稳定性分数,这个流程还能输出每个氨基酸残基的摇摆幅度(RMSF)。这就等于给了一张「结构风险地图」。pLDDT 分数低于 50 的区域,通常 RMSF 会超过 5 埃,说明这些地方非常不稳定。而 pLDDT 高于 90 的核心区域,则非常刚性,RMSF 可能小于 2 埃。
对于做蛋白质工程的人来说,这就是一份明确的改造指南。你可以直接对着这张图说:「看,这个柔性 loop 是个麻烦,我们在这里引入一个脯氨酸增加刚性,或者设计一个二硫键把它锁住。」
一个重要的提醒:物理化学常识不能丢。
当然,这个方法也不是万能的。论文里提到了一个很有意思的反例:一个蛋白的 pLDDT 分数很高(约 85),但在模拟中却几乎散架了(RMSD 高达 11.7 埃)。
为什么?因为研究者在建立模拟体系时,遗漏了对结构至关重要的化学信息,比如没有明确定义二硫键,或者缺少了必要的金属离子、辅因子。这给我们敲响了警钟:AI 模型和物理模拟都是工具,它们无法弥补你化学知识上的短板。如果你的模拟设置从一开始就错了,那结果必然是「垃圾进,垃圾出」。
AlphaUnfold 提供了一个连接 AI 预测和物理现实的桥梁。它不是最终答案,而是一个高效的过滤器。在投入昂贵的实验室资源之前,先用这种方法快速「压」一下你的蛋白设计,筛掉那些看起来很美但实际上不堪一击的候选者,能帮我们省下大量的时间和金钱。
📜Title: AlphaUnfold: Probing Potential Unfolding and Structural Fragility in AlphaFold3 Models via Short-Time High-Pressure MD
🌐Paper: https://www.biorxiv.org/content/10.64898/2026.04.22.720259
💻Code: https://github.com/pegados/pipeline_AlphaUnfold
3. 用几个例子就能预测抗体性质?这个新 AI 模型搞定了批次效应
做实验的都清楚,批次效应 (batch effects) 是个多大的麻烦。同一批样品,今天测和明天测,结果就可能因为仪器状态、试剂批次漂移一点。这种系统性偏差不大不小,但足以让你的数据分析和模型预测一团糟。现在,亚马逊的研究者们拿出了一个叫 CA-MAP 的框架,思路很有意思,它不试图消除批次效应,而是学会「理解」它。
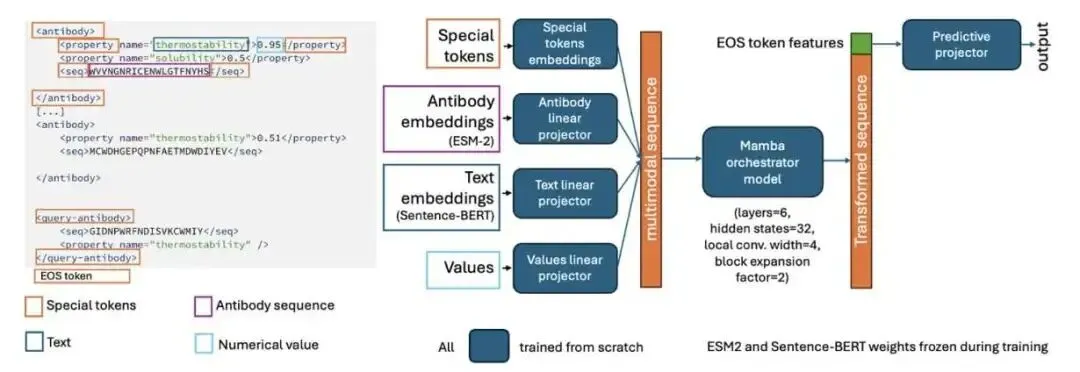
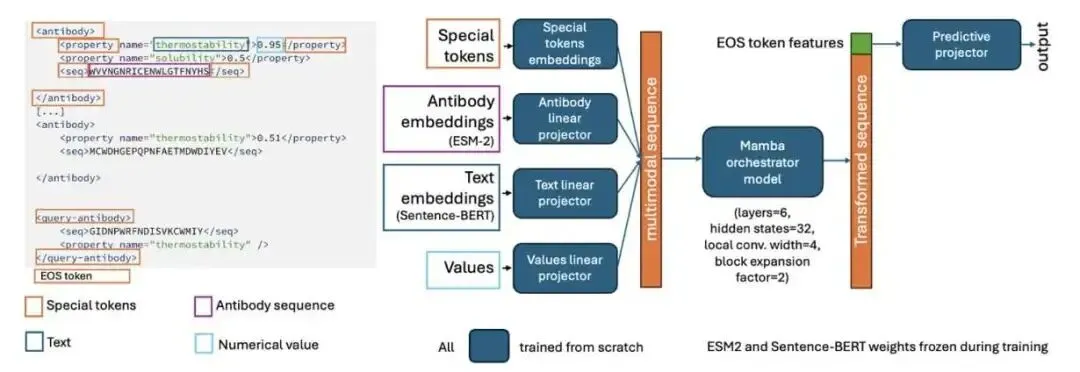
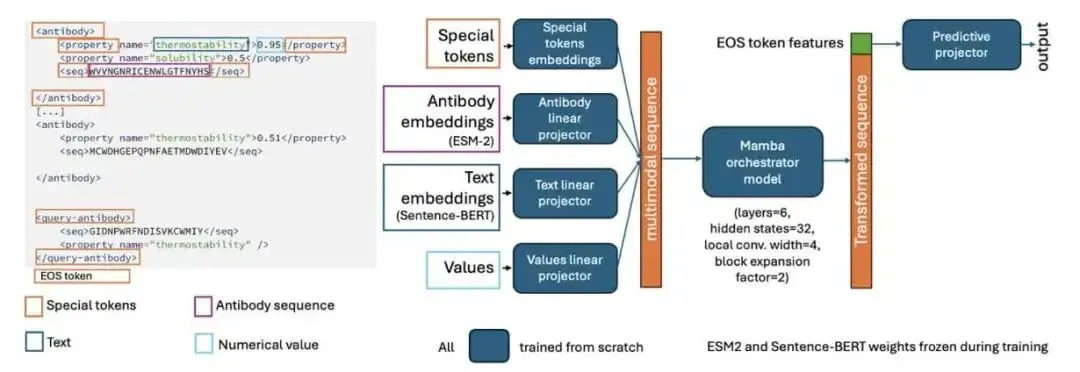
它的工作原理很像我们带新人。你不会把新人抓去从头到尾重新培训一遍。你会直接把他带到仪器前,给他看几个标准品,告诉他:「看,今天这台机器上,样本 A 的读数是 1.2,样本 B 是 1.4」。新人看懂了这个「上下文」,就能准确测出未知样本 C 的数值了。
CA-MAP 做的事情一模一样。你给它一个包含几个已知抗体序列及其性质(比如疏水性、溶解度)的「提示」(prompt),然后再给它一个需要预测的目标序列。模型会分析这些例子,找出其中的规律——包括隐藏的批次效应——然后对目标序列给出预测。这种能力叫做「上下文学习」(in-context learning)。
为了让模型真正学会从上下文中找线索,而不是偷懒只看目标序列本身(这叫「捷径学习」),研究者用了一个很方法,叫 AB-context-aware 训练。他们在训练时,会给每一组样本的性质数值偷偷加上一个随机的、隐藏的「变换」。这样一来,模型如果只看单个序列,完全无法得到正确答案。它必须对比上下文中的所有样本,才能反推出这个隐藏的变换是什么,然后才能正确预测目标。这就好比故意把仪器的刻度弄偏,逼着你必须用标准品来校准一样。
效果立竿见影。当研究者人为地给测试数据加入 0 到 0.3 的加性偏差来模拟批次效应时,常规微调的大模型 Tx-Gemma 预测疏水性的相关性系数 (Spearman's ρ) 直接从接近 1 崩到了 0.58。而 CA-MAP 几乎不受影响,性能稳定在 0.99 左右。它确实从上下文中看懂了批次效应并修正了它。
在模型架构上,他们没有选择那种把蛋白质序列和文字描述强行编码到同一个空间里的「大一统」方案。那样做很容易丢失信息。他们的做法是,蛋白质序列用 ESM-2 编码,性质名称用 Sentence-BERT 编码,数值也单独编码,各走各的路。然后,用一个非常小巧的 Mamba 模型作为「指挥家」(orchestrator),来整合这些不同来源的信息。Mamba 是一种状态空间模型,处理长序列上下文的效率很高。整个 CA-MAP 模型总参数量约 1.25 亿(主要是冻结的 ESM-2 和 BERT),但需要训练的参数只有区区 18.2 万,训练和推理都很快。
更让人惊讶的是它的泛化能力。CA-MAP 甚至能预测它在训练中从未见过的性质。比如,研究者在训练时完全排除了免疫原性和正电荷异质性这两个性质。但在预测时,只要在上下文中提供几个包含这两个性质的例子,模型就能举一反三,给出相当准确的预测(相关性系数能从 0.2 左右提升到 0.73)。这说明模型不只是死记硬背,而是在学习不同理化性质之间更底层的关联。
当然,这个工作也有局限。目前用的性质标签主要来自计算,还没有用大量的湿实验数据验证。另外,它也没有测试抗体最重要的功能——亲和力。但无论如何,CA-MAP 展示的思路非常亮眼:让模型学会在上下文中动态适应,而不是试图用一个静态模型解决所有问题。这对于处理充满噪音和变化的真实世界生物数据,可能是一条正确的路。
📜Title: Context-aware multi-property antibody predictor: a novel framework integrating text and protein language models
🌐Paper: https://doi.org/10.1038/s41540-026-00723-1
💻Code: https://github.com/amazon-science/ca-map/
4. AI 当审稿人:自动验证生物学代码,准确率超 90%
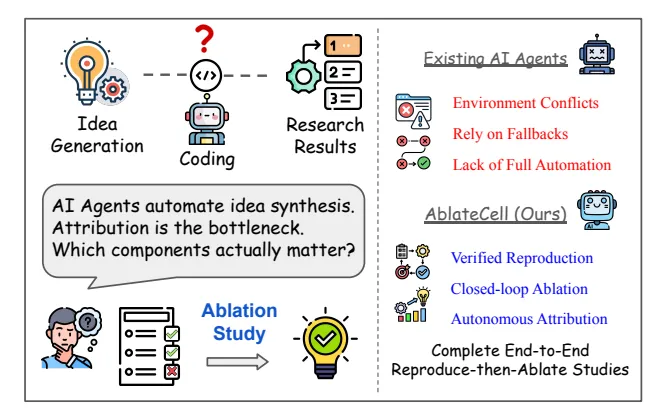
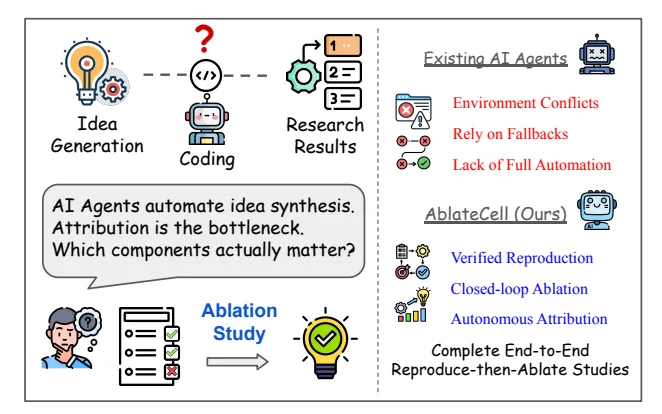
做计算生物学研究,我们都遇到过一个头疼的问题:想复现一篇论文里的模型,结果发现代码库的环境配不齐、数据路径不对、脚本跑不通。这个过程能耗掉几周甚至几个月的时间,最后你可能都不知道这个模型到底是不是像论文里说得那么好。
现在,有人开发了一个叫 AblateCell 的 AI 智能体,就是来解决这个问题的。你可以把它想象成一个极其耐心和严谨的计算生物学博士后,它能 7x24 小时地帮你干两件核心的脏活累活:复现代码和分析模型。
第一步:像侦探一样复现代码 (Reproduce)
AblateCell 的工作流程很直接。首先,它会拿到一个 AI 虚拟细胞(AI Virtual Cell, AIVC)模型的代码库。这些代码库通常很复杂,数据集格式特殊(比如 h5ad),训练和评估脚本也耦合得很紧,复现起来很麻烦。
AblateCell 会在一个隔离的 Docker 环境里启动工作。它通过一个「规划 - 执行」的流程,自动配置环境、解决依赖问题、修正数据路径、找到正确的入口脚本,然后从头开始重新运行官方的训练和推理流程。整个过程的产出都是可验证的,包括能跑通的脚本、模型检查点、性能指标和详细日志。它把最繁琐的环境配置和 Debug 工作自动化了。
第二步:像做实验一样拆解模型 (Ablate)
成功复现出基线结果后,更有意思的部分来了。AblateCell 开始进行「消融研究」(ablation study)。
消融研究是模型开发里的一个标准操作,就是通过系统性地移除或替换模型的某个部分,来观察性能会下降多少,从而判断这个部分的重要性。手动做这件事很枯燥,而且容易出错。
AblateCell 把这个过程变成了一个基于图的自动化执行流程。它会把代码库的各个组件(比如一个特定的编码器、一个损失函数)看作是可变异的节点。然后,它会开始进行一系列受控的「代码库突变」:关闭某个功能、替换某个模块、或者缩放某个参数。
这里的核心算法很聪明。在计算资源(比如 GPU 小时)有限的情况下,想找到最重要的组件,本质上是一个信息增益问题。AblateCell 采用了一种多臂老虎机策略来平衡探索和利用。
打个比方,这就像你在一个赌场里,面前有一排老虎机(模型的各个组件),但你的筹码(计算时间)有限。你不会在每台机器上都花光筹码,而是会先在每台机器上试几把。如果某台机器(某个组件)看起来很有「潜力」(移除它会导致性能大幅下降),你就会投入更多筹码去重点验证它。
它的奖励函数设计得也很实在:r(x) = |Δ(x)| − λ · cost(x)。这个公式的意思是,它寻找的是那种能引起巨大性能变化 |Δ(x)|,同时测试成本 cost(x) 又不高的组件。这完全符合科研实验室的实际需求:用最少的资源找到最关键的发现。
为了保证每个消融实验干净、独立,互不干扰,AblateCell 还用了一个很巧妙的工程技巧:Git worktrees。每次实验都在一个独立的 Git 工作区里进行,相当于为每个实验都复制了一份干净的代码库。这样既能安全地并行测试,又能完整地追溯每一次修改和结果。
效果怎么样?
研究者在 CPA、GEARS 和 BioLORD 这三个主流的单细胞扰动预测代码库上测试了 AblateCell。结果相当亮眼。
它还发现了一些符合领域知识的结论。比如,在 GEARS 模型中,它发现图神经网络(GNN)编码器是绝对的核心,拿掉它之后,模型的均方误差(MSE)能飙升近 90%。在 CPA 模型里,统一的隐空间嵌入影响最大。这些结论不是靠研究员猜出来的,而是 AblateCell 自动、定量地跑出来的。
这个工具的价值在于,它不仅仅是让复现工作变简单了。它带来了一种更严谨、更自动化的科学验证方法。它能帮助我们快速地甄别一个新模型里,哪些是真正的创新,哪些只是「炼丹」技巧。这对于加速药物研发、提高整个领域研究的可信度,意义重大。
📜Title: AblateCell: A Reproduce-then-Ablate Agent for Virtual Cell Repositories
🌐Paper: https://arxiv.org/abs/2604.19606

5. 弃用微调:LLM 让蛋白设计成功率翻倍
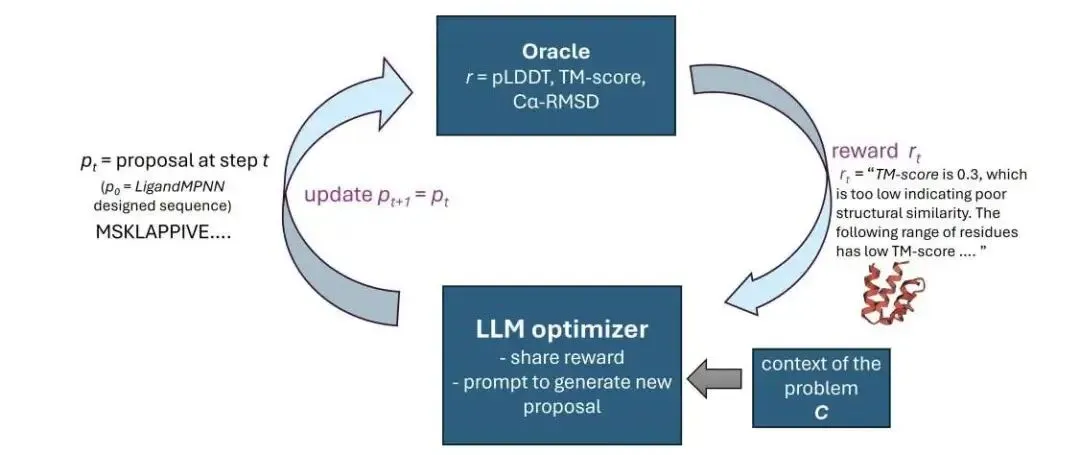
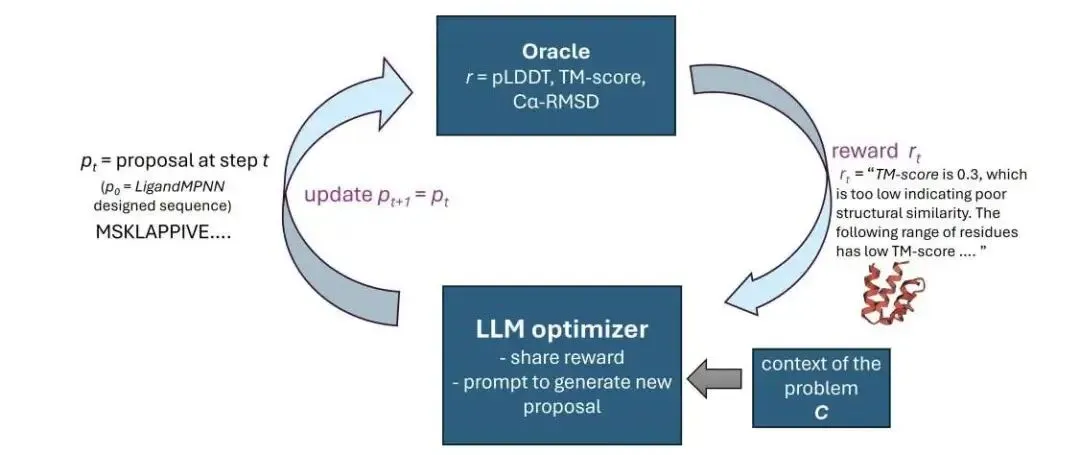
做蛋白设计的人都知道,用 LigandMPNN 跑出来的序列,丢进结构预测软件里经常惨不忍睹。碰到这种情况,以前只能瞎蒙突变或者干脆重新生成。这篇论文提出了 RosettaSearch 框架。它的思路直接击中了痛点:利用大语言模型(LLM)在推理阶段做序列优化。研究人员压根没有去微调模型,全靠一套反馈闭环。
这是它的工作原理。系统把大语言模型当成生成式优化器。它给模型喂两类信息:一是全局的结构评价分数,包含置信度(ss-pLDDT)、全局相似度(TM-score)和几何偏差(Cα-RMSD);二是局部的残基注释。系统会指明:「第 20 到 25 位氨基酸的预测置信度低,偏差大。」模型接收这些反馈后,就会给出针对性的氨基酸替换建议。
这种基于反馈的编辑管用吗?数据摆在眼前。在近 400 个具有挑战性的 PDB 单体重新设计案例中,起始序列来自表现一般的 LigandMPNN。经过 RosettaSearch 优化后,结构保真度全面提升,设计成功率直接增加了约 2.5 倍。研究者专门做了一个对照实验。如果只在这些报错的区域做算力对等的随机突变,结果往往更糟。这就证明了成功率的提升实打实来自于大语言模型对反馈信息的理解,它确实做出了符合化学和结构逻辑的替换。
搜索策略决定了优化的效率。逐个修改序列容易走弯路。RosettaSearch 采用了一种基于优先级的并行搜索策略。这就像我们在做化合物库筛选。系统维护一个候选序列池,反复挑出排名前 K 的序列,并行提出 K 个新的修改方案,交给 RosettaFold3 验证,再把所有结果放回池子里。这样做防止了模型过早卡在局部最优解。
优化过程中遇到了一个经典的机器学习问题:模型为了拿高分开始作弊。大语言模型会耍小聪明,生成连续的丙氨酸、重复特定的基序,甚至篡改序列长度。为了阻止这种行为,研究者在文本提示词里加入了软约束。他们要求模型检查 6-mer 重复率、遵守长度限制,并在有配体的情况下死死固定住结合位点的氨基酸,以保住功能性相互作用。
自己出卷自己考容易掩盖问题。为了验证这些序列真的靠谱,研究者把最终结果交给了独立的结构预测工具 Chai-1 进行评估。结果依然稳健。这说明模型学到了真实的折叠规律,绕开了 RosettaFold3 的特定偏好。这个方法也适用于从头设计的骨架。在 RFDiffusion 生成的骨架上,哪怕不提供参考序列,RosettaSearch 依然把成功率从 72.4% 拉升到了 89.5%。
论文最后做了一个多模态的前沿尝试。他们直接把预测结构的渲染图喂给视觉语言模型。模型看完 3D 结构的二维截图后,能给出空间感更强的推理过程。现阶段这部分的效果和纯文本优化持平,但这显然给未来的计算辅助设计指明了一个新方向。
📜Title: RosettaSearch: Multi-Objective Inference-Time Search for Protein Sequence Design
🌐Paper: https://arxiv.org/abs/2604.17175

