 夜雨聆风
夜雨聆风
特别提示
今天这期内容对追求高分和清晰分析路线的老师们来说非常友好!“单细胞扰动预测模型评测+构建Snakemake评测框架+整合六个单细胞扰动数据集”,思路清楚,逻辑完整,主角是我们熟悉的“方法评测”,拿下9分+,快来看看吧!
如果内容对大家有帮助,麻烦点个一键三连,大家的支持就是我们创作的动力,谢谢!

文章题目: scArchon:单细胞扰动预测模型的可复现评测框架
期刊: Genome Biology
影响因子:9.4
研究思路: 构建Snakemake评测框架+整合六个单细胞扰动数据集+复现九类扰动预测工具+统一可视化和统计指标+加入DEG/通路等生物学指标+比较线性与控制基线+评估跨物种泛化和消融鲁棒性+输出可复用的容器化评测流程
优势: 方法评测,系统比较单细胞扰动预测模型在统计指标和生物学保真度上的差异
发表年月:2026年05月
─── 这篇文章为什么值得看? ───
这篇文章的价值不只在于围绕单细胞扰动预测模型评测展开分析,而在于把“单细胞扰动预测模型评测+构建Snakemake评测框架+整合六个单细胞扰动数据集”组织成了一条相对完整的研究路线。 结果部分重点围绕scArchon建立了统一的单细胞扰动预测评测设计、降维图会影响模型表现的直观判断、一些深度模型并不稳定优于线性或控制基线逐步展开。
─── 研究背景 ───
【背景知识】:
单细胞转录组数据让研究者可以在细胞类型、患者和物种层面观察药物、感染或刺激等扰动后的表达变化。近几年,很多深度学习模型开始尝试根据未扰动细胞预测扰动后的转录状态,这类方法如果可靠,理论上可以用于药物反应建模和个体化响应预测。但这一领域的比较长期比较分散:不同论文往往只和少数工具比较,使用的数据集、降维图和评价指标也不统一,导致模型到底是在捕捉真实扰动信号,还是只在某些指标上看起来好,并不容易判断。
【研究目的】:
本研究提出scArchon这一可复现、模块化的评测平台,用统一流程评估单细胞扰动响应预测工具。作者希望回答三个关键问题:不同工具在统一数据和统一指标下表现如何;常用的全局统计指标是否足以反映生物学保真度;当任务变成患者差异、跨物种迁移或数据规模变化时,现有模型的稳健性边界在哪里。
─── 研究思路 ───
【思路流程图】:
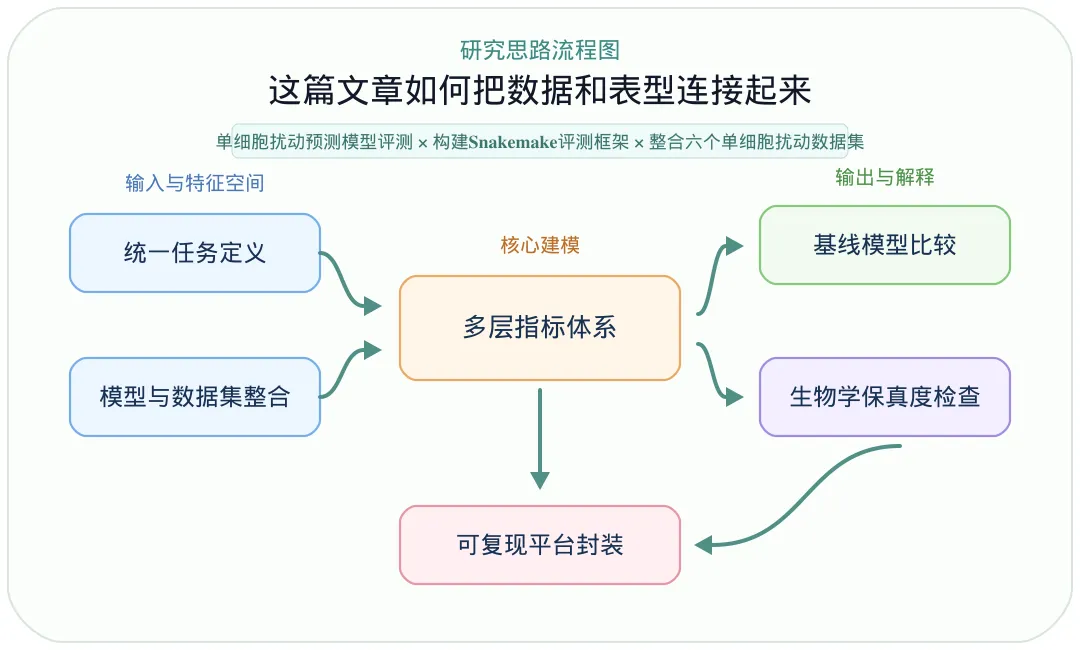
【本研究采用的关键分析策略】:
·统一任务定义: 将任务定义为从控制状态细胞预测扰动后状态,并采用留一协变量交叉验证,让模型在未见过的细胞类型、患者或物种上接受评估。
·模型与数据集整合: 评测trVAE、scDisInFact、scVIDR、scPRAM、scGen、SCREEN、CPA、scPreGAN和CellOT,并纳入Kang、H. Poly、Nault、Species、Glioblastoma和Interferon alpha等六个单细胞扰动数据集。
·多层指标体系: 同时使用PCA/UMAP/t-SNE可视化、MSE、Wasserstein distance、t-test、R2、DEG重叠、富集通路重叠和语义相似度等指标,避免只依赖单一分数。
·基线模型比较: 将深度学习工具与线性扰动模型、未扰动控制基线进行比较,判断复杂模型是否真正超过简单假设。
·生物学保真度检查: 进一步分析预测DEG和富集GO/通路是否与真实扰动一致,用来识别可能的虚假生物学信号。
·可复现平台封装: 用Snakemake和Docker/Singularity容器管理工具运行环境,输出预测矩阵、评估表格和可视化结果,便于后续加入新模型或新数据集。
─── 研究结果 ───
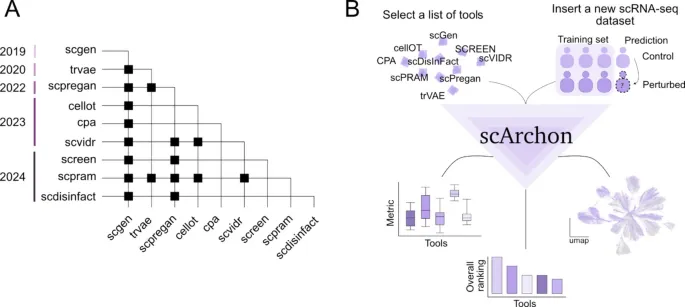
1. scArchon建立了统一的单细胞扰动预测评测设计
这部分想回答的问题: 单细胞扰动预测工具众多,但不同研究之间缺少统一任务、统一数据和统一指标,导致模型比较不够透明。
作者怎么做: 作者构建scArchon平台,选择九个代表性工具,在六个公开单细胞RNA-seq扰动数据集上进行留一协变量评测,并用Snakemake和容器化环境保证可复现运行。
主要结果:
· scArchon可以从输入的h5ad数据开始,依次执行模型训练、扰动状态预测、结果保存、指标计算和可视化。
· 评测任务覆盖细胞类型、患者、物种等不同协变量,强调模型对未见群体的泛化能力。
· 平台不仅输出模型预测结果,也输出多种统计指标、生物学指标和降维可视化。
可以怎么理解: scArchon把原本分散的单细胞扰动模型比较,整理成了一套可复现、可扩展的评测流程。
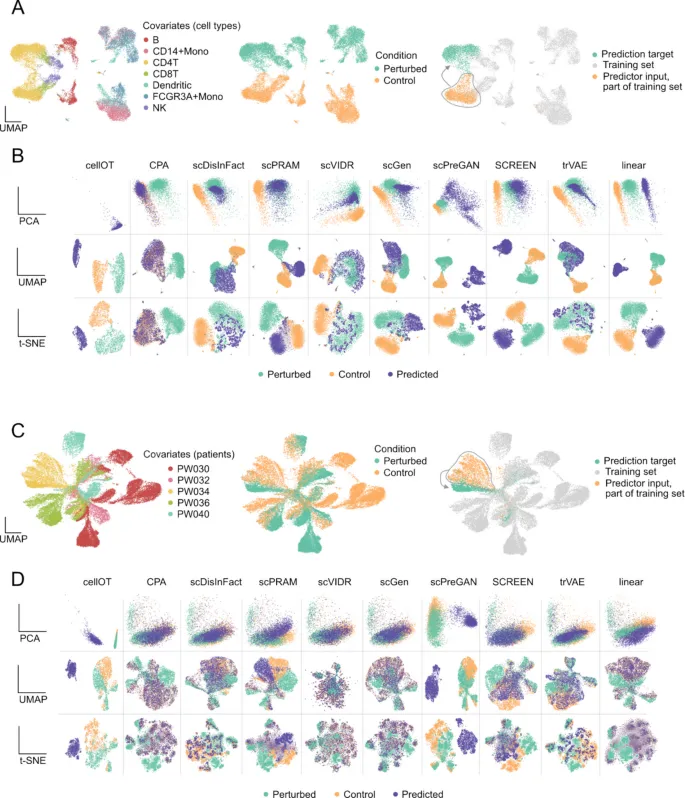
2. 降维图会影响模型表现的直观判断
这部分想回答的问题: 很多模型论文会用PCA、UMAP或t-SNE展示预测细胞和真实扰动细胞是否重叠,但不同降维方式是否会给出不同结论?
作者怎么做: 作者在Kang数据集和胶质母细胞瘤数据集中,对多个工具的预测结果分别用PCA、UMAP和t-SNE展示,并比较控制、扰动和预测细胞的空间关系。
主要结果:
· 在Kang数据集的PCA图中,scPRAM、scDisInFact、scGen、scVIDR和trVAE的预测细胞更接近真实扰动状态。
· 切换到UMAP后,部分在PCA中看似重叠良好的工具出现分离,例如scPRAM和scVIDR的表现会变弱。
· 在更复杂的胶质母细胞瘤数据中,即便部分工具在简单数据集表现较好,也更容易把预测细胞拉回控制组附近。
可以怎么理解: 低维可视化不能单独作为扰动预测模型好坏的判断依据,同一结果在不同降维方式下可能呈现不同印象。
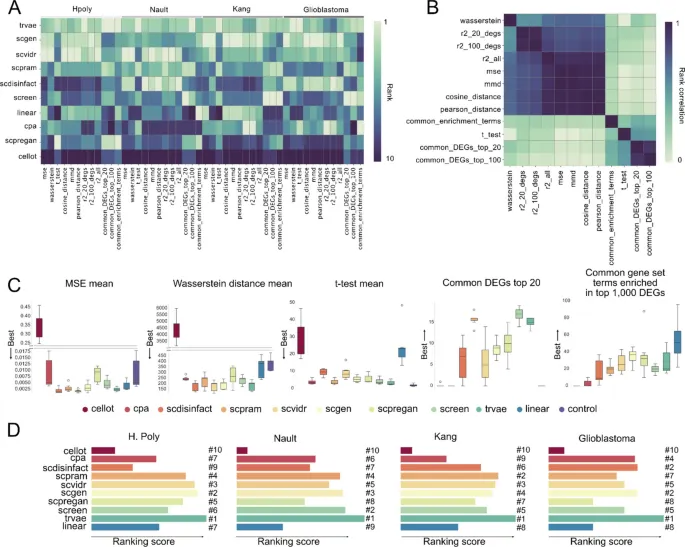
3. 一些深度模型并不稳定优于线性或控制基线
这部分想回答的问题: 在统一统计指标和生物学指标下,复杂深度学习工具是否稳定超过简单基线?
作者怎么做: 作者综合R2、Wasserstein distance、t-test、top DEGs重叠和富集通路重叠等五类相对独立指标,对模型在多个数据集和预测折中的表现进行排名。
主要结果:
· trVAE在总体综合排名中表现最好,scGen、scPRAM和scVIDR也在不同数据集中较为稳定。
· CellOT、CPA和scPreGAN在部分任务中可能低于线性模型,说明复杂模型并不必然优于简单扰动假设。
· 不同数据集之间模型表现差异明显,例如scDisInFact在部分数据集排名较低,但在胶质母细胞瘤任务中表现较好。
· 某些工具在全局统计指标上得分尚可,但DEG或通路层面的生物学信号保留较弱。
可以怎么理解: 单细胞扰动预测模型的排名高度依赖数据集和评价指标,复杂模型需要和简单基线一起评估。
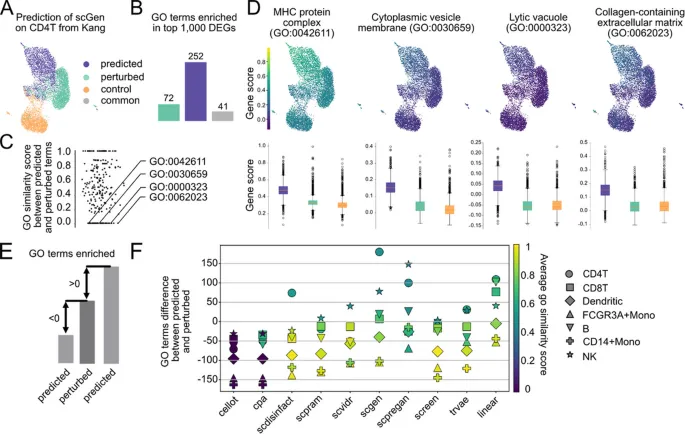
4. 生物学指标揭示预测模型可能产生虚假扰动信号
这部分想回答的问题: 如果一个模型在全局指标上表现不错,它预测出的通路和基因层面变化是否一定可信?
作者怎么做: 作者比较预测细胞和真实扰动细胞之间的GO富集项重叠,并进一步计算预测GO术语与真实扰动GO术语之间的语义相似度。
主要结果:
· 某些模型会预测出远多于真实扰动条件的富集GO terms,导致表面上看起来有较多重叠。
· 对语义相似度为零的预测GO terms进一步检查后,发现部分预测状态中相关基因表达被人为抬高,可能产生虚假富集。
· 作者将这种现象类比为扰动模型中的“biological hallucinations”:模型生成了看似有生物学意义、但并不对应真实扰动结构的信号。
可以怎么理解: 评估扰动预测模型不能只看全局距离或富集项数量,还需要检查基因层面和通路语义是否真正贴近真实扰动。
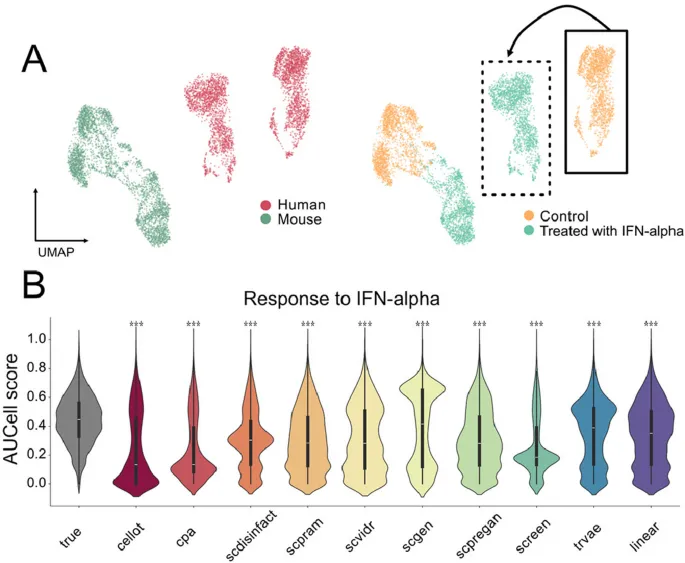
5. 跨物种扰动迁移仍是现有工具的明显短板
这部分想回答的问题: 模型能否把一种物种中学到的扰动响应迁移到另一种物种,尤其是从小鼠推到人类细胞?
作者怎么做: 作者先在包含小鼠、兔、猪、鼠等多物种LPS刺激数据上进行留一物种评测,再在小鼠和人T细胞干扰素α刺激数据中测试跨物种预测,并用AUCell评估干扰素响应通路活性。
主要结果:
· 在多物种LPS任务中,工具没有产生有意义的低维重叠,线性模型在某些MSE指标上反而排名较高。
· 在小鼠到人T细胞的干扰素α任务中,各工具预测的通路活性分布与真实人类扰动细胞显著不同。
· 这说明即便单个基因层面看似合理,模型也可能无法保留跨物种扰动响应的高阶调控结构。
可以怎么理解: 现有单细胞扰动预测工具在跨物种泛化上仍然有限,不能轻易把动物模型中的扰动响应外推到人类细胞。
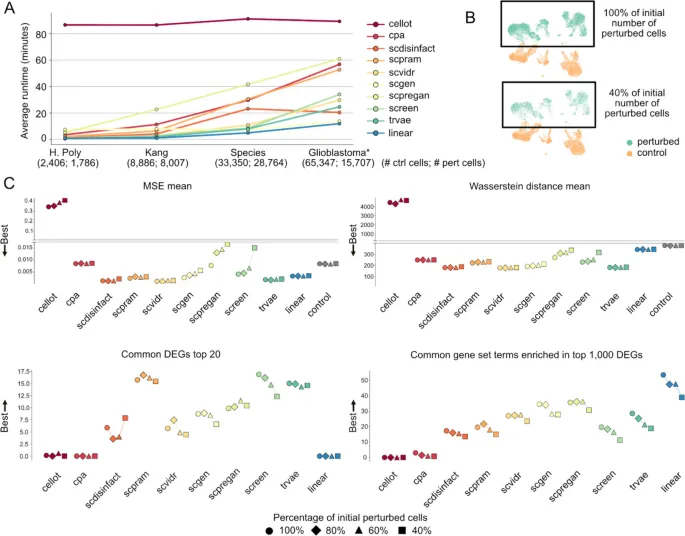
6. 运行成本和消融实验显示不同工具的稳健性边界
这部分想回答的问题: 这些工具在运行成本、训练样本减少和基因集变化时是否仍然稳定?
作者怎么做: 作者比较不同工具在多数据集上的运行时间,并在Kang数据集中逐步减少扰动细胞比例,在胶质母细胞瘤数据中减少输入基因数量,观察指标变化。
主要结果:
· scPreGAN、scDisInFact、scPRAM和scVIDR运行较快,CellOT因为缺少GPU支持而耗时最高。
· 当扰动细胞减少到80%、60%和40%时,多数工具的MSE和Wasserstein指标相对稳定,但部分生物学指标会下降。
· 减少高变基因数量后,Wasserstein distance可能改善,但富集项和DEG重叠等生物学信号仍会受影响。
· scArchon本身依赖CUDA兼容环境、Docker/Singularity镜像和一定存储空间,实际使用时需要考虑计算资源。
可以怎么理解: 模型评测不仅要看最终分数,还要同时考虑运行成本、输入数据变化和生物学信号恢复的稳定性。
─── 总结 ───
整体来看,这篇发表于 Genome Biology 的文章围绕 单细胞扰动预测模型评测 展开。 它的核心路线可以概括为:单细胞扰动预测模型评测、构建Snakemake评测框架、整合六个单细胞扰动数据集、复现九类扰动预测工具。 结果部分则通过scArchon建立了统一的单细胞扰动预测评测…、降维图会影响模型表现的直观判断、一些深度模型并不稳定优于线性或控制基线等模块逐步支撑主线。 这类研究的重点不是给模型排一个固定榜单,而是把评测任务、数据集选择、简单基线、统计指标、生物学指标和计算成本放在同一个框架下比较。这样得到的结论更接近方法边界本身:哪些模型在特定任务中相对稳健,哪些指标可能放大表面优势,哪些预测信号还需要回到基因和通路层面核对。对于单细胞扰动预测模型评测这样的方向,统一评测的作用是暴露模型能力和生物学保真度之间的差距,而不是把某个分数当成最终答案。
后话
这篇文章更适合作为“单细胞扰动预测模型评测该怎么被严肃评测”的方法学案例。它提醒我们,模型分数需要和简单基线、生物学指标、跨数据集泛化以及运行成本放在一起看。
─── 参考文献 ───
· scArchon:单细胞扰动预测模型的可复现评测框架. Genome Biology. DOI: 10.1186/s13059-026-04104-z.
