 夜雨聆风
夜雨聆风【导读】
两个月前,我在做一篇文献解读的时候就写了:后续我会专门写一篇AI+催化剂设计的方法论总结。当时觉得,四篇论文读下来,思路已经七七八八了,整理一下应该不难。
真动笔才发现,难的不是整理,而是取舍。哪些坑是共性的、哪些只是个案,哪些建议对所有人都适用、哪些只适用于特定场景,这些边界远比我想象的模糊。我尽量把自己能想到的都写出来了,但认知有限,总结不到位的地方肯定不少。如有偏差,望大家指正,也欢迎讨论。
AI建模的上限,不是算法决定的
先抛一个核心观点,AI构效关系建模的上限,是由化学信息的编码深度与数据质量决定的,算法只是逼近这个上限的工具。
这句话的意思是:你喂给AI的原料决定了它能做出什么菜,算法只是火候。原料是烂菜叶,米其林大厨也炒不出好味道。
接下来,我把整个流程拆成六步,咱们一步步聊。
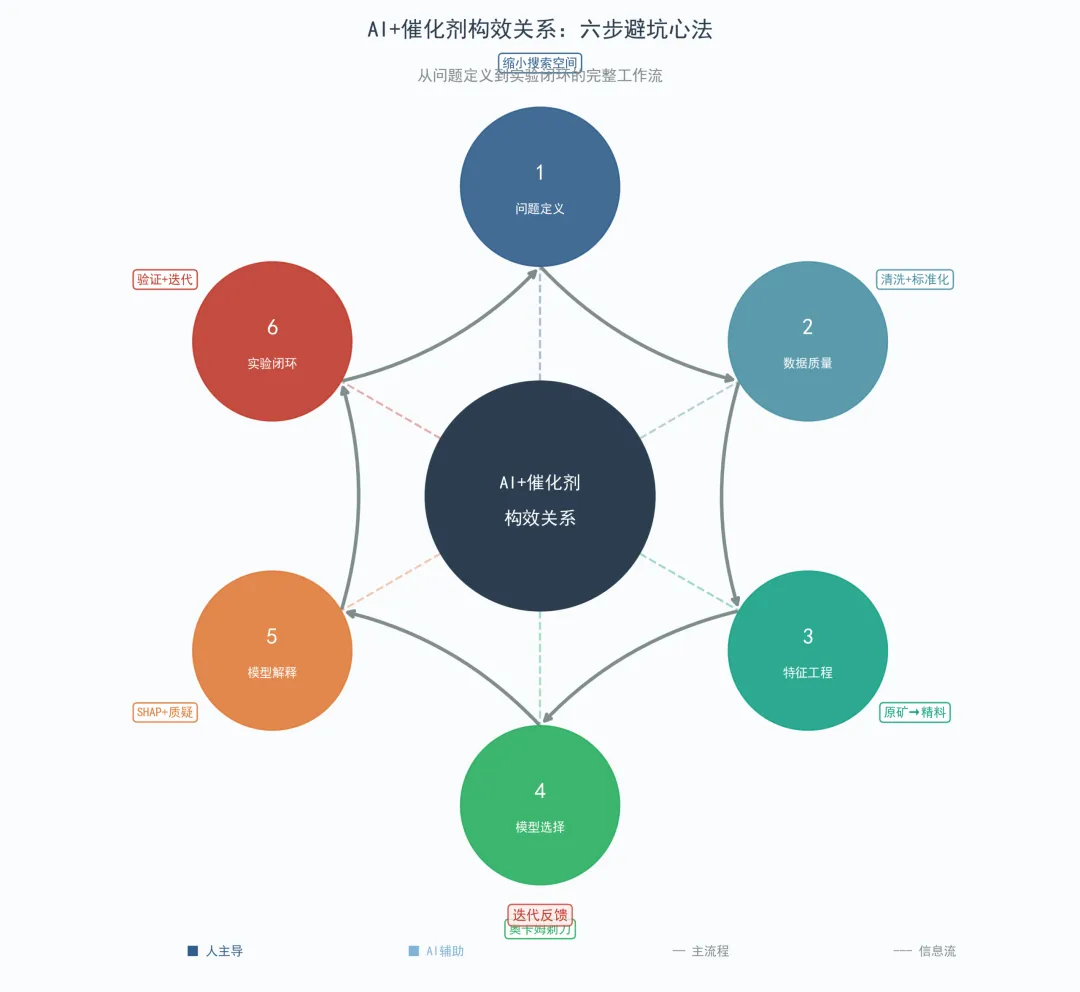
第一步:问题定义——你到底想让AI帮你干嘛?
做实验前我们需要知道目标产物是什么,做AI也一样。别一上来就说我要预测催化剂性能,我们需要把这句话翻译成AI能懂的数学语言。
我们是要预测一个具体性能数值,还是只需要判断不同种类催化剂的好/坏?后者是分类问题前者是回归问题。我们关注的性能指标有多少个,多个性能指标之间如何权衡,哪些是关键指标,哪些是次要指标。这些定义直接决定了后续每一步的走向。
例如:我之前读的一篇发在Science Advances上的OER电催化论文,作者做的就很有分寸。他们没有让AI直接预测过电位,而是先通过数据挖掘提炼出Ru必须占一席、Sr/Mn/Zn是优质掺杂元素这些先验规律,再在这个有边界的探索空间里让主动学习迭代。这就是问题定义的价值:先缩小搜索空间,再让AI干活。
第二步:数据获取与质量控制——你的原料干净吗?
AI建模中有一句老话,Garbage in, garbage out。数据质量是决定模型生死的第一关。
这里的一个大坑就是迷信文献里的大数据库。把不同课题组、不同装置测的数据混在一起直接训练,表面看样本量上去了,实际上模型学到的可能不是构效关系,而是这个数据点长得像哪个实验室的作品。
我读的那一篇发在npj Computational Materials上的CO₂加氢论文,就用组内相关系数量化不同来源数据的批次效应。结果很震撼——69.8%的性能波动来自实验室差异,不是催化剂本身。作者还做了一个验证:拿高温烧结做基准测试,不控制批次效应时,In₂O₃体系居然表现出高温处理后比表面积显著增加的荒谬结论,控制后才回到合理区间。
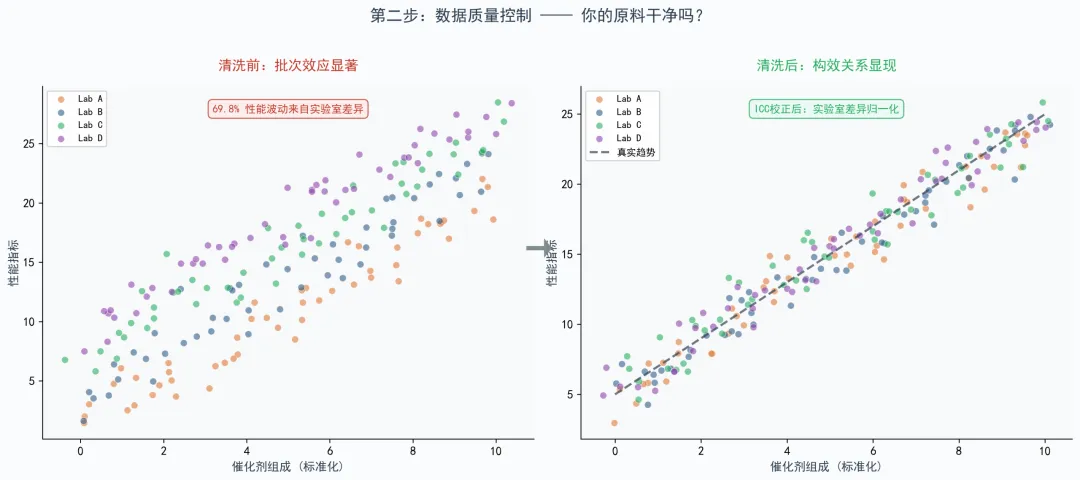
这说明什么?说明AI并不懂化学,它只擅长捕捉数据里的稳定模式。而实验室习惯,恰恰也是一种稳定模式。
如果没有条件做大批量实验怎么办?这块我仍然认为可以参考开源数据库或者文献里面的实验数据,但是要结合自己的少批量实验数据,有选择性的适用。实验条件有限的情况下我们需要花费更多时间在数据清洗和标准化上面。
第三步:特征工程——你喂的是原矿还是精料?
这一步是拉开建模上限的核心步骤。输入的变量有没有包含真实的物理化学意义,决定了AI能不能学到真正的构效关系。
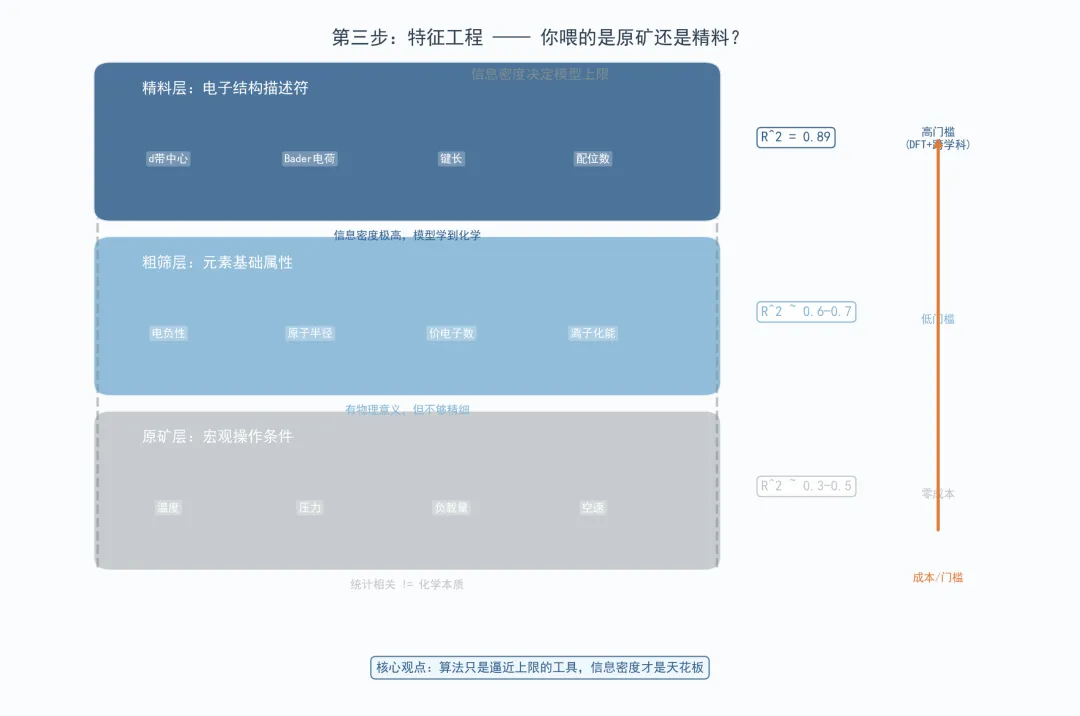
这块的问题在于,我们做实验中只能观测到宏观操作条件-温度、压力、负载量,因此把这些当输入特征,但其实更深层次的是我们改变宏观的操作条件影响催化剂本身的化学性质。就像我之前读的一篇CO₂-ODHP论文,作者直接把元素含量作为特征输入模型,这个模型只能在统计层面做粗筛。
进阶的做法是引入化学描述符。例如:另一篇发在Advanced Science上的甲烷氧化论文,作者用DFT计算了d带中心、Bader电荷、键长等电子结构描述符,信息密度极高,模型R²直接冲到0.89。但门槛也高,要求研究者同时精通DFT、机器学习和催化理论,这样的人才至少也要是高年级的博士或者在企业相关领域工作多年的硕士。
但是如果没有算力怎么办?最简单的思路,找低成本替代。元素周期表基础属性——电负性、原子半径、价电子数——虽然粗糙,但比纯宏观条件强。或者寻求做理论计算的外部合作,君子性非异也,善假于物也。
第四步:模型选择与训练——奥卡姆剃刀原则
算法只是逼近上限的工具。不要陷入越复杂越好的算法崇拜,适合数据体量和特征的才是最好的。
如果实验数据就几十条,但是在用深度学习,这叫拿大炮打蚊子。小数据场景下,传统的机器学习模型随机森林、XGBoost就差不多了。我之前读的几篇论文,270样本用随机森林、92样本用XGBoost,都是合理选择。关键不是模型多炫酷,而是这个模型是否合适我要解决问题的场景,。
例如:那篇甲烷氧化用的就是XGBoost模型+超参数优化,也取得很好的效果。工具没有绝对好坏,关键在于是否针对问题特点做了合理适配。把省下来的时间花在特征工程上,回报更高。
第五步:模型解释与因果验证
我有一个特殊的观点,跑出一个R²=0.9的模型不能完全称为胜利,能解释为什么,且符合化学直觉,甚至能纠正人类偏见的模型,才有价值。
这里其实有一个问题就是把相关性当成因果性。AI说加了某种助剂转化率高,可能只是因为加了助剂的实验恰好都在高温下做的。那篇CO₂加氢论文的作者就追问了一句:SHAP反映的到底是因果性还是相关性?这显示出对方法局限性的清醒认知。
所以一定要用SHAP等工具看看模型到底在关注什么特征。如果SHAP给出的重要特征违背了基本化学常识,先回去查数据是不是有坑。关键是要有质疑模型的习惯。哪怕不做完整的因果推断,至少问自己三个问题:这个特征重要性符合化学直觉吗?有没有变量没考虑?如果换一个相似体系,结论还成立吗?
第六步:实验闭环与迭代——纸上得来终觉浅
模型给出的最优催化剂,必须在反应釜里验明正身。AI不是取代实验,而是给实验指路。很多工作做到预测就结束,模型和实验之间是断层的。那篇OER论文的价值,恰恰在于它把闭环做完整了:数据挖掘给方向、主动学习迭代配方、实验验证性能、DFT解释机理,最后还在真实PEM电解槽单电池里测到了高电流密度。
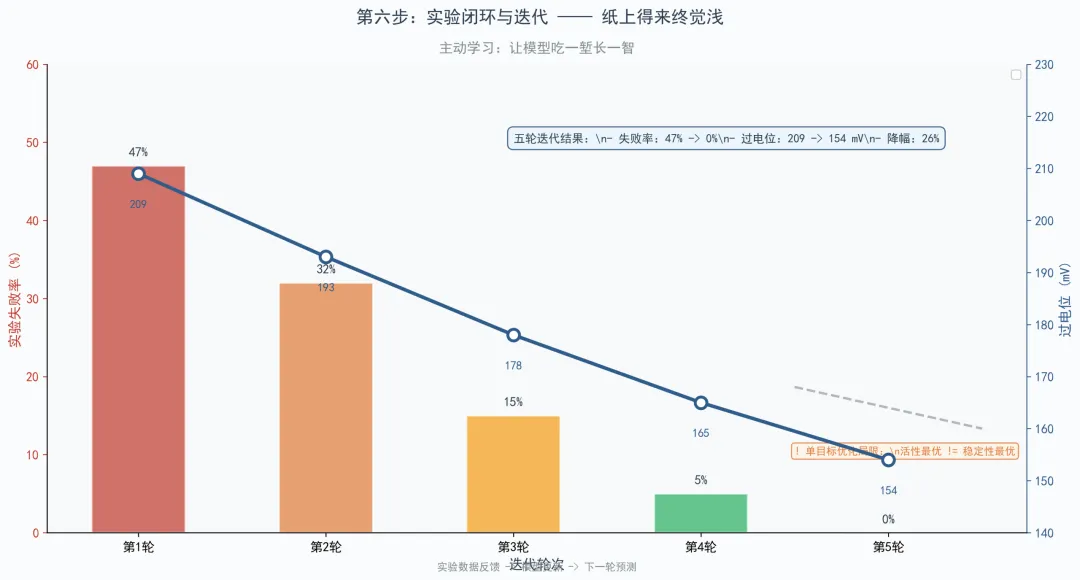
主动学习的核心思想是把实验验证的结果——包括失败的数据——重新喂给模型,让它吃一堑长一智。那篇论文五轮迭代,失败率从47%降到0%,过电位从209 mV降到154 mV,这就是闭环的力量。
但这里也有局限。那篇论文的主动学习只优化了活性,稳定性没纳入目标函数,结果半电池活性最好的样品,在单电池和稳定性测试里反而不如另一个样品。这说明化学化工的问题往往是多目标优化问题,单目标的预测模型可能只是玩具。如果进行多目标优化,寻找两个目标的帕累托最优解,可能是更好的方式。
如果实验条件有限,至少做到模型预测后手动验证几个关键点,把验证结果反馈回来修正模型。哪怕只迭代一轮,也比预测完就结束强得多。
给资源有限同仁的极简避坑路线
收拢一下。不要迷信算法,多花时间在数据清洗和特征思考上。给没有大团队、没有超算的同仁三条落地建议:
第一,数据。尽量保证数据的来源一致干净,且有良好的空间分布。
第二,特征。考虑更深层次的化学,哪怕用最基础的元素属性,也比纯宏观条件强。
第三,心态。把AI当成一个懂统计学的高级实验员,而不是能凭空变出诺贝尔奖成果的魔法。
总结
从四篇论文的零散启发,到这篇方法论的成型,中间隔了两个月。不是不想写,是越写越发现,AI+催化这个交叉领域,是一个太过宏大的课题,我现在的总结充其量是一张局部地图,不是全球导航。
下一步,我会找一个具体场景,把这套方法论跑一遍,代码开源。如果你也相信AI在化工的未来就在不久的将来,欢迎关注,我们一起用AI重构化工经验。
我是Wiggins,一个在化工厂里敲代码的工程师。
真正有价值的技术,往往不在聚光灯下,而在那些愿意俯下身,把泥土和枯枝利用起来的人手里。
参考文献 (References)
[1] Liu, H., Liu, K., Zhu, H., Guo, W., & Li, Y. Explainable machine-learning predictions for catalysts in CO2-assisted propane oxidative dehydrogenation. RSC Advances, 14(10), 7276-7282
[2]Xu, W., Bai, L., Qi, J., Sun, X., Wang, B., Zhao, Z., & Li, B. A natural language processing to causality framework for robust knowledge extraction of CO₂ hydrogenation with batch effect control. npj Computational Materials.
[3] Ding, R., Liu, J., Hua, K., Wang, X., Zhang, X., Shao, M., Chen, Y., & Chen, J. Leveraging data mining, active learning, and domain adaptation for efficient discovery of advanced oxygen evolution electrocatalysts. Science Advances, 11(14).
[4]Pan, M., Zhang, T., Dong, J., Xie, Y., Zhong, K., Guan, W., Wei, H., Fu, C., & Su, Y. High-Throughput Screening and Interpretable Machine Learning for Rational Design of Bimetallic Catalysts for Methane Activation. Advanced Science.
