 夜雨聆风
夜雨聆风Emyx:一个“小而美”的全原子蛋白生成模型,在计算酶设计基准上实现新突破
在AI制药与计算生物学领域,计算酶设计是极具前景但挑战巨大的方向。其核心任务之一,是围绕一个已知的催化活性位点(motif),生成一个能够稳定支撑该位点并容纳配体的全新蛋白质骨架(scaffold)。这要求生成模型不仅要有极高的几何精度,还要能产生结构多样的设计方案。然而,当前主流全原子生成模型通常沿袭自结构预测领域的复杂架构,导致训练成本高昂,且生成的骨架多样性有限。
近日,来自英国生物技术公司Xyme的研究团队提出了一种名为 Emyx 的高效全原子蛋白质结构生成模型。这项研究通过简化架构、引入创新的训练与采样策略,在权威的AME(Atom-level Motif Scaffolding)酶设计基准测试中,以更小的参数量和更低的训练成本,实现了比当前先进模型(如RFdiffusion3和Proteína-Complexa)更高的成功率,并生成了结构更新颖、更多样化的蛋白质骨架。
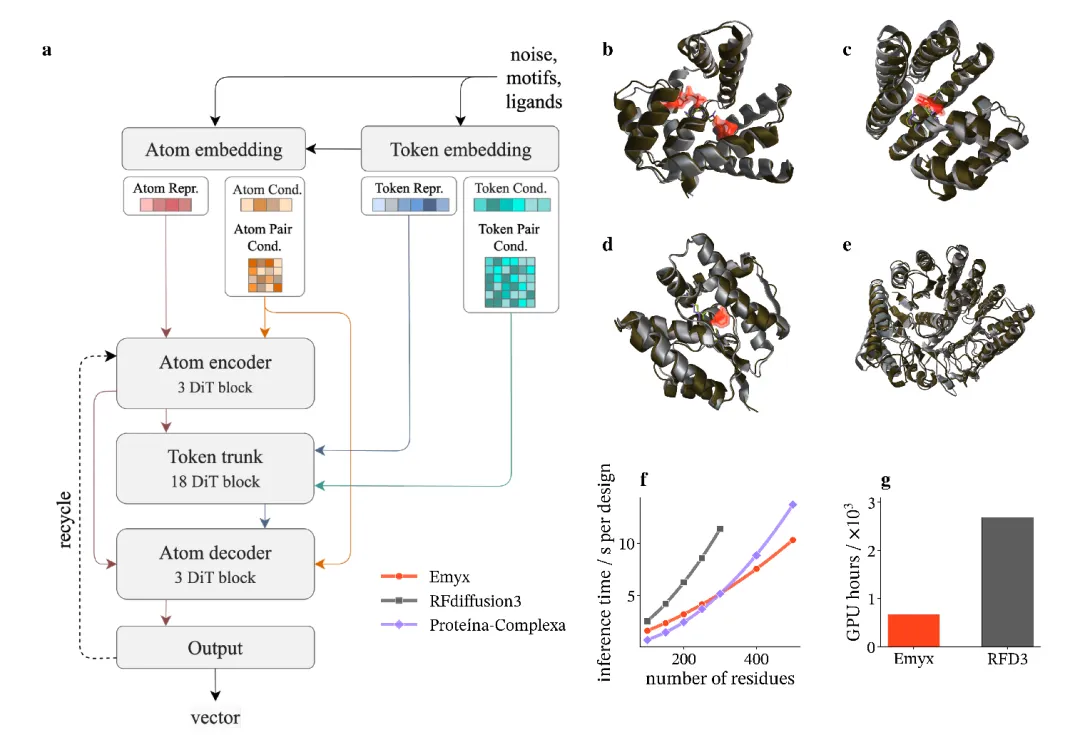
图 1:Emyx模型架构概览
一、 背景:从骨架到全原子生成的挑战
早期的深度学习方法(如RFDiffusion)主要在蛋白质骨架层面进行生成,随后需要借助逆折叠模型(如ProteinMPNN)来获取序列。这种方法的局限在于,催化残基必须被预先指定在序列的特定位置,这降低了生成的成功率并增加了计算复杂度。更近期的模型(如RFDiffusion3, Proteína-Complexa)通过支持“非索引”的活性位点支架,让模型自行决定催化残基在链上的位置,从而大幅提升了成功率。
然而,这些先进模型依然背负着从结构预测领域继承而来的“昂贵”架构,依赖于密集的成对表示和复杂的嵌入层。这些架构带来了巨大的内存开销,限制了模型的可扩展性。研究团队提出了一个核心观点:对于生成模型而言,这种复杂性可能是不必要的。
结构预测模型的输入是丰富的序列上下文信息(如多序列比对、同源模板、蛋白质语言模型嵌入),这些信息为初始表示注入了丰富的语义。而生成模型的条件输入主要是几何约束(如链连接性、活性位点和配体几何),这些信息在开始时是“贫瘠”的,其关系结构需要在信息跨令牌混合后才得以建立。因此,对于生成器,架构的简洁性可能更为有利。
二、 核心方法:轻量架构与高效流匹配
Emyx是一个基于条件流匹配(Conditional Flow Matching)的全原子蛋白质结构生成模型。其核心创新在于,将模型能力几乎完全集中在标准的Transformer模块中,摒弃了从结构预测继承的昂贵架构。
1. 高效的原子-token双级表示:Emyx采用统一的Rep14表示法,每个token(残基或配体原子)由固定大小的14个原子槽位矩阵表示。通过轻量级的原子级Transformer块编码局部原子细节,再通过门控交叉注意力池化为令牌级嵌入。一个深层的Transformer主干(含稀疏自注意力和学习到的成对偏置)负责捕获长程结构上下文,最后再通过交叉注意力将token上下文转换回原子,进行最终的原子级解码。
图 2:严格sc-RMSD评估示例
2. 流匹配训练与EDM重参数化:Emyx使用线性插值进行流匹配训练,这比扩散模型中常用的方差保持计划具有更低的训练梯度方差和更快的收敛速度。更重要的是,研究团队推导出将流匹配插值精确重参数化为EDM噪声水平框架的公式。这是一个与模型无关的映射,使得Emyx无需重新训练即可使用为扩散模型设计的先进采样方法(如Karras计划和随机扰动),从而将流匹配的训练效率与最先进的扩散采样技术相结合。
三、 关键结果:更准、更新、更快
研究团队在包含41个催化活性位点的AME基准上对Emyx进行了全面评估,并与RFdiffusion3和Proteína-Complexa进行了对比。评估采用了严格的自我一致性协议:生成骨架→用LigandMPNN重新设计序列→用Boltz-2重新预测结构。
1. 提出更严格的评估标准:研究指出,AME原先使用的“重原子sc-RMSD”标准可能高估了成功率(约2倍),因为它只检查局部活性位点的几何恢复,而未验证整体折叠的一致性。为此,他们提出了“严格sc-RMSD”标准,要求同时满足:全局骨架RMSD < 2.0 Å(确保整体折叠恢复)和活性位点尖端原子RMSD < 1.5 Å(确保催化几何准确)。
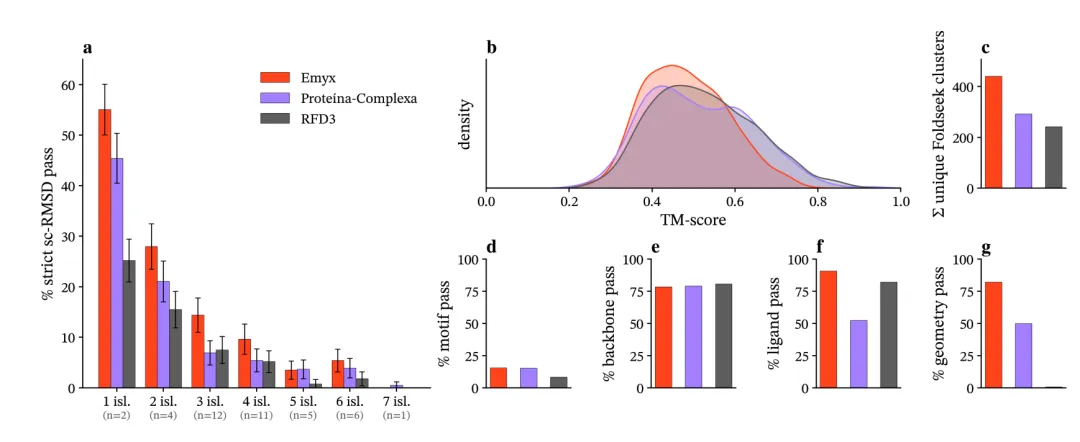
图 3:在严格sc-RMSD标准下,Emyx在AME基准上的成功率全面领先
2. 性能全面领先:在严格sc-RMSD标准下,仅1.4亿参数的Emyx取得了13.4%的成功率,解决了39/41个靶点,显著优于Proteína-Complexa(8.8%)和RFdiffusion3(6.7%)。同时,Emyx成功设计的骨架与已知PDB结构的相似性最低(中位TM-score 0.48),且产生的独特结构簇数量最多(441个),表明其生成的结构新颖性和多样性更优。
3. 效率优势显著:Emyx的训练仅需682 GPU小时(8块H200 GPU上约3.55天),比RFdiffusion3(约2688 GPU小时)减少了约4倍。在单块A10G GPU上进行推理时,Emyx的速度也快于RFdiffusion3,且序列越长优势越明显。
4. EDM采样提升效果:通过前述的重参数化使用EDM采样器,比使用标准的SDE Euler-Maruyama积分获得了更高的成功率,证实了将扩散采样创新直接迁移到流匹配生成器的有效性。
四、 行业意义与展望
从AIDD和计算酶设计的视角看,Emyx的研究提供了有价值的启示:针对生成任务特性进行架构简化,而非盲目继承预测模型的复杂设计,可以同时实现性能提升与效率优化。其提出的严格sc-RMSD评估标准,也有助于领域更准确地衡量模型的实际设计能力。
这项工作的价值在于,它降低了计算酶设计的门槛。更低的训练成本和更快的推理速度,使得更多资源有限的研究机构能够参与前沿探索。这对于推动AI制药在工业生物催化、药物合成及绿色化学等领域的应用具有积极意义。
当然,Emyx仍有改进空间,例如目前仍需独立的逆折叠步骤进行序列重设计,未来的方向是开发联合序列-结构生成模型。此外,自我一致性评估流程本身计算成本较高,也是大规模比较的瓶颈。
总 结
Xyme团队开发的Emyx模型,通过聚焦Transformer核心能力、采用高效流匹配训练及创新的EDM重参数化采样,在计算酶设计的关键任务上实现了“更准、更新、更快”的突破。它不仅以更小的模型在权威基准上取得了当前最佳性能,其简洁高效的设计理念也为蛋白质设计和AI制药领域的模型开发提供了新的思路。随着此类技术的不断成熟,我们有望看到更多具有新颖功能的人造酶被高效设计出来,加速生物医药与合成生物学的创新进程。
【文章原文】
https://arxiv.org/pdf/2606.19377
