 夜雨聆风
夜雨聆风





高熵合金结构建模的补充说明及相关软件的安装
上一篇文提供了ATAT构建四元合金的一个典型算例,执行起来是非常快的。但是在实际高熵合金体系,建模的计算量是非常大的,举个例子Ti0.2Zr0.31Hf0.09Nb0.2Ta0.2:
可以选择Ta.cif(体心立方),也可以采用Ni.cif(面心立方)来建模,具体需要以实验测量结构作为参考,此处以Ni.cif面心立方结构作为参考,转换得到的Ni.in文件为:
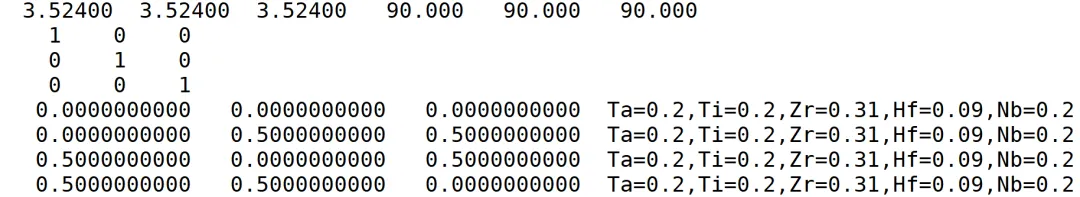
可以看到参考结构包含4个原子坐标,高熵合金包含5种元素,其中Zr为0.31,Hf为0.09,其它为0.2。那么,从超胞的角度看,应该需要4n个原子,从元素的角度来看,应该需要100m个原子(m和n均为整数)。
那么,要同时满足这两个条件的最小整数是400。
此时,执行mcsqs -n 400 -l=Ni.in由于原子数特别多,需要改用并行的命令执行(需要加载intel环境变量):
mpirun -np 20 mcsqs -n 400 -l=Ni.in即便并行运算,也需要较长时间(超过24小时),Objective_function逐步收敛到-1过程也需要很长时间。可以在mpirun -np 20 mcsqs -n 400 -l=Ni.in命令的基础上添加mpirun -np 20 mcsqs -n 400 -l=Ni.in -ms=15000,限定最大运行15000次,即使Objective_function没有达到Perfect_match,也停止运行了。或者在运行过程中,查看mcsqs.log中Objective_function,是否达到了期望的收敛值,按Ctrl+c可随时取消任务(要达到Perfect_match,需要的增大超胞,尽量达到Perfect_match)。
当然,还有一种方法就是将Ti0.2Zr0.31Hf0.09Nb0.2Ta0.2近似为Ti0.2Zr0.3Hf0.1Nb0.2Ta0.2,这样最少40个原子就可以模拟了(但仍需要增大胞中原子个数,避免原子偏析)。
图1 包含400个原子的Ti0.2Zr0.31Hf0.09Nb0.2Ta0.2结构
另外,在执行过程,可以跳过mcsqs -n 32 -l=Ni.in,直接手动新建sqscell.out文件定义超胞,直接执行mpirun -np 20 mcsqs -l=Ni.in -rc,利用设定超胞和蒙特卡洛算法,通过并行直接生成高熵合金结构。
附相关软件的安装教程:
竟然有人说找不到ATAT的下载链接,为了放置链接地址的域名变化,特详细给出检索过程:
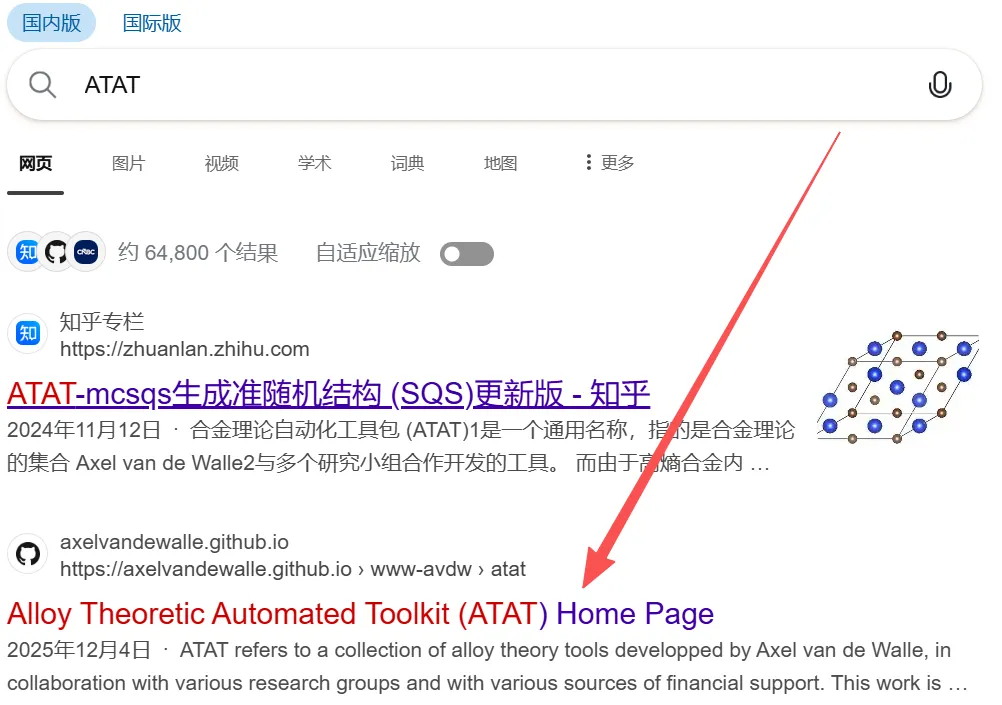
搜索结果第二条就是官网,进入官网后,找到Download,通常直接选择最新的whole toolkit(Stable version)就可以了。

下载安装包:atat3_50.tar.gz
安装步骤为:
1、上传服务器,解压:
tar -zxvf atat3_50.tar.gz
2、加载intel编译器的环境变量(每个机器略有不同),修改makefile文件:
cd atat
vi makefile
其中CXX=g++,MPICXX=mpicxx -DATAT_MPI,其它内容不动,然后执行make命令,然后make install,诸多二进制文件被自动拷贝到$HOME/bin中,包括corrdump、kmesh、genstr、gensqs、mcsqs。自此,ATAT就安装好了。
ATOMKIT就更容易安装了,在sourceforge下载代码后,直接
tar -zxvf atomkit.0.9.0.linux.x64.tar.gz
解压,将其中atomkit可执行文件移动到$HOME/bin就可以了。
欢迎使用qvasp软件:
[1] W. Yi, G. Tang, et al.qvasp: A Flexible Toolkit for VASP Users in Materials Simulations, Comput. Phys. Commun., 2020, 257, 107535.
关注科研牛,科研不迷路
长期分享可靠的科研方法
