 夜雨聆风
夜雨聆风





让AI读懂分子与病历,华东交大DMAPLM如何用“注意力”挖掘潜在救命药?为药物重定位提供生物学解释
DMAPLM: A multimodal pretrained framework for computational drug repositioning

近期推送
原文摘要
药物再利用为现有药物发现新的治疗适应症提供了一条有效途径。然而,当前的计算药物再利用模型常常面临数据稀缺、异质性等挑战,因此普遍适用性有限。为了解决这些限制,本研究引入了DMAPLM,一个用于预测药物-疾病关联的多模态预训练框架,以进一步进行药物再利用筛选。DMAPLM采用轻量级双编码器架构,使用ChemBERTa-2对药物SMILES字符串进行分子编码,使用BioB-BERT对多字段疾病文本进行语义编码。该框架通过对比学习明确对齐药物和疾病表示,并采用注意力加权池化强调信息丰富的分子子结构。最终使用随机森林分类器基于增强的多模态特征进行关联预测。我们汇编了一个全新的综合基准数据集以评估性能。广泛实验表明,DMAPLM显著优于六种最先进的基线模型,在五折交叉验证下实现了0.8919的AUROC和0.9116的AUPR,提升了高达9%。此外,DMAPLM在具有挑战性的冷启动场景中表现出稳健的性能,突显了其识别新药物-疾病关系的实际效用。案例研究以及分子对接分析证实了我们的预测的可解释性和生物学意义。我们的研究为计算药物重定位提供了一种强大且可解释的方法。
原文解读
研究背景
-
研究问题:这篇文章要解决的问题是如何有效地重新利用现有药物来发现新的治疗适应症。现有的计算药物重定位模型通常面临数据稀缺、异质性以及泛化能力有限的问题。
-
研究难点:该问题的研究难点包括:数据稀缺和异质性导致的模型泛化能力差;复杂计算模型的可解释性不足;多模态数据的整合和分析技术挑战。
-
相关工作:该问题的研究相关工作包括基于网络传播、传统机器学习和深度学习的药物重定位方法。最近的研究还利用大型语言模型(LLMs)如BioBERT和ChemBERTa进行药物重定位。
研究方法
这篇论文提出了DMAPLM,一种用于计算药物重定位的多模态预训练框架。
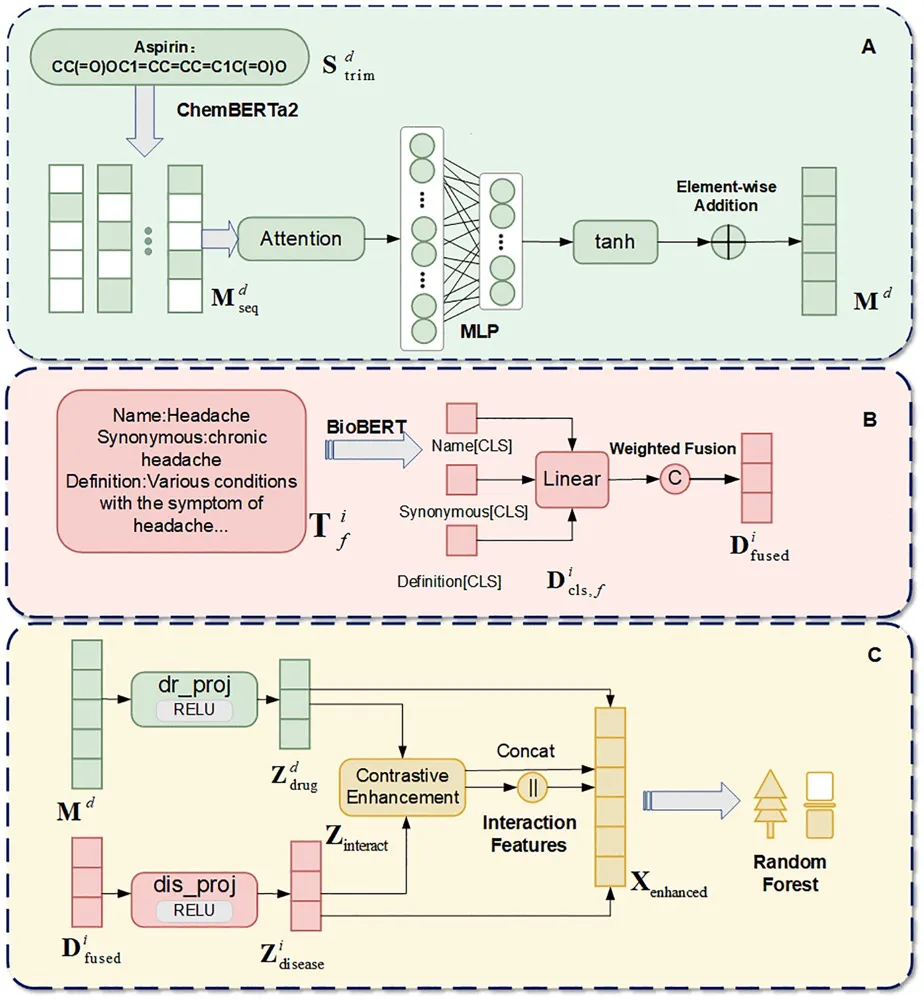
具体来说,
-
分子编码:使用ChemBERTa-2对药物SMILES字符串进行编码。ChemBERTa-2是一种基于RoBERTa的预训练语言模型,专门用于分子表示学习。SMILES字符串首先被截断到最大长度512,然后通过ChemBERTa-2转换为上下文化嵌入。
-
疾病文本编码:使用BioBERT对多字段疾病文本进行编码。BioBERT是一种领域特定的预训练语言模型,适用于生物医学文本挖掘。从DrugMAP 2.0数据库中提取疾病名称、同义词和定义三个文本字段,分别进行编码并通过加权聚合融合。
-
注意力加权池化:对分子嵌入进行注意力加权池化,以聚合序列信息。注意力池化过程如下:
-
对比学习增强特征:通过对比学习框架将嵌入投影到一个共享的潜在空间,增强特征表示。对比学习过程如下:
-
随机森林分类:基于增强的多模态特征,使用随机森林进行分类预测。随机森林通过自助抽样和多数投票聚合多个决策树的预测结果。
实验设计
-
数据集构建:从DrugMAP 2.0数据库中搜索并收集实验确认的药物-疾病关联数据,构建一个包含1455种疾病、2622种药物和5993个药物-疾病关联的基准数据集,关联密度为0.0016(0.16%)。
-
实验设置:已知药物-疾病关联标记为正样本,未知关联标记为负样本。采用五折交叉验证(5-CV)进行性能评估,每个样本在训练和测试中各使用一次。
-
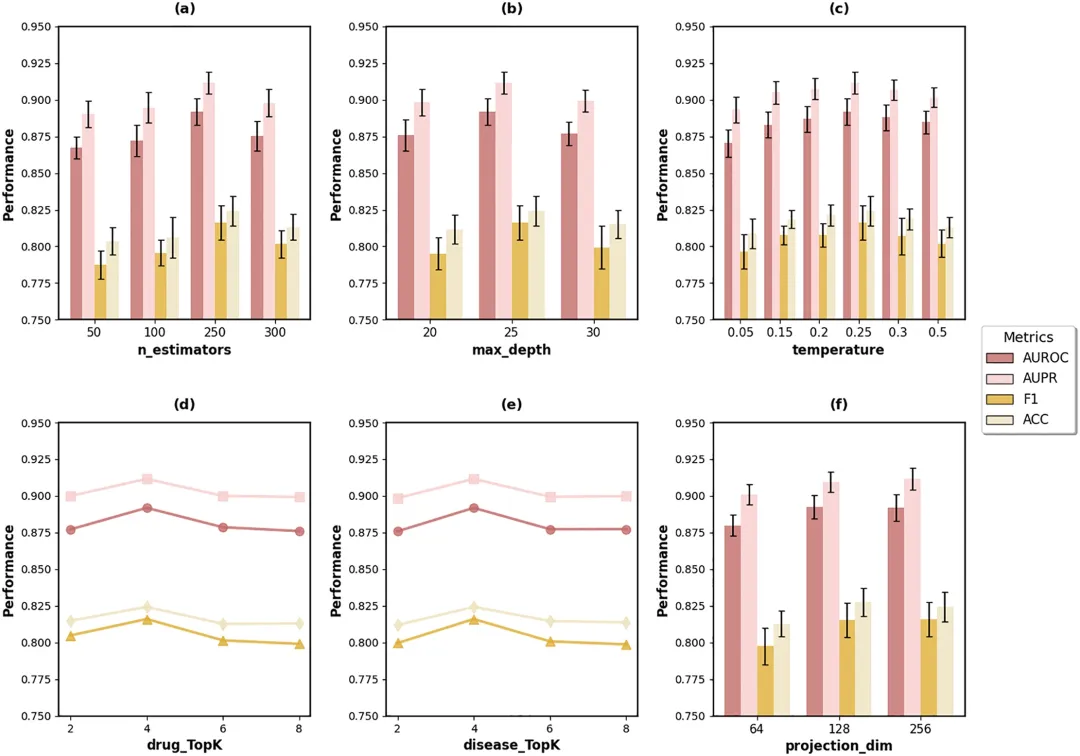
超参数配置:通过网格搜索评估不同超参数设置对预测性能的影响,关键超参数包括随机森林的估计器数量、最大深度、温度、药物TopK、疾病TopK和投影维度。
结果与分析
-
性能评估:在五折交叉验证和冷启动设置下,DMAPLM在AUROC和AUPR方面比最先进的六种基线模型提高了最多9%。具体结果为AUROC 0.8919和AUPR 0.9116。
-
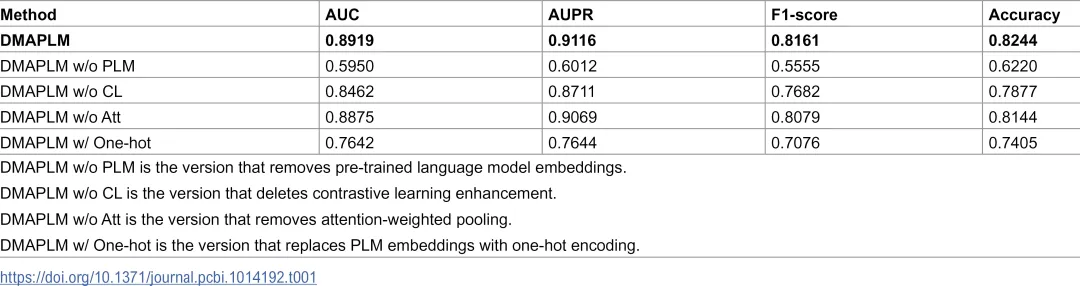
消融测试:通过消融测试评估不同组件对DMAPLM性能的贡献。移除预训练语言模型嵌入导致性能显著下降,移除对比学习导致性能下降,移除注意力加权池化也导致性能下降。
-
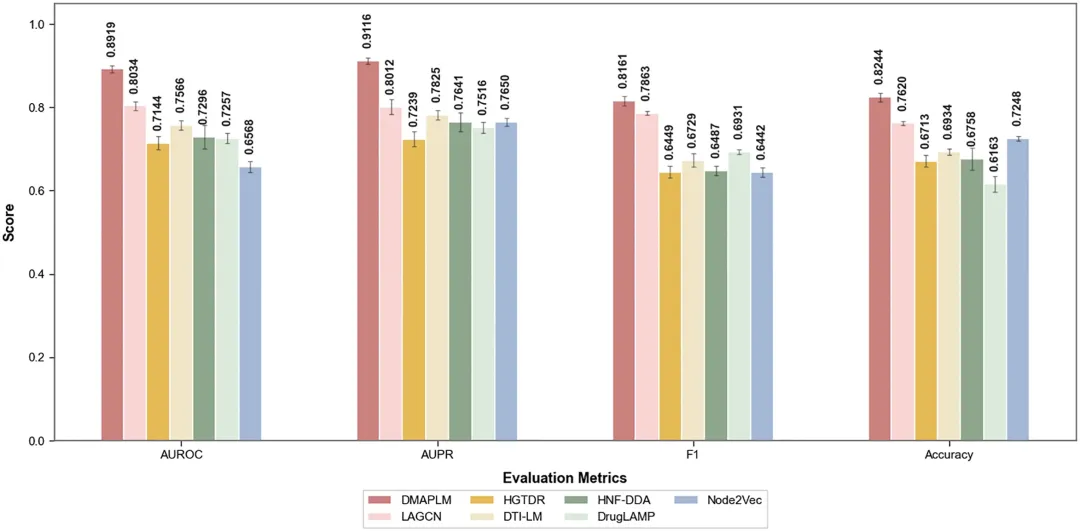
模型比较:DMAPLM在所有评估指标上均优于其他竞争模型,特别是在AUROC和AUPR方面显著领先。
-
冷启动预测:在C1、C2和C3三种冷启动场景下,DMAPLM在所有场景中都表现出色,特别是在最具挑战性的C3双冷启动场景中,DMAPLM仍保持稳定的预测性能。
-
Top-K评估:DMAPLM在所有Top-K指标上均表现优异,特别是在Top-1预测中,P@1达到0.8139,比第二好的LAGCN高出3.73%。
-
鲁棒性分析:通过引入高斯噪声评估DMAPLM对数据质量问题的鲁棒性,结果表明DMAPLM在中等噪声水平(20%)下仍能保持强性能,即使在严重噪声(50%)下也能保持合理的预测能力。
-
特征质量评估:对比学习显著提高了嵌入空间的判别能力,正样本相似性达到0.679,负样本相似性降至0.146,Cohen’s d值达到4.56。
总体结论
DMAPLM提供了一种有效且可解释的多模态框架,用于计算药物重定位。通过结合预训练语言模型和对比表示对齐,DMAPLM在预测准确性和泛化能力方面取得了显著提升。案例研究和分子对接分析进一步证实了DMAPLM生成的生物合理假设的翻译潜力。总体而言,DMAPLM是一个可扩展且稳健的计算工具,可用于加速药物重定位,其与实验验证管道的集成有望促进更有效的治疗发现。
特点评价
-
轻量级跨模态架构:提出了一种基于预训练语言模型的双重编码框架,利用ChemBERTa-2和BioBERT分别对药物分子和多场疾病文本进行编码,无需构建复杂的图结构。
-
显式跨模态对齐:通过对比学习显式对齐分子结构和疾病文本语义空间,并通过注意力机制捕捉互补特征,增强跨模态判别能力。
-
鲁棒性和可解释性:框架在数据稀疏和冷启动场景下表现稳定,具有生物学上可解释的预测,适合实际临床应用。
-
综合基准数据集:通过搜索DrugMAP 2.0数据库,构建了一个包含1,455种疾病、2,622种药物和5,993个药物-疾病关联的综合基准数据集。
-
对比学习增强特征提取:引入对比学习框架,将嵌入投影到一个共享的潜在空间,通过InfoNCE对比损失优化投影网络。
-
随机森林分类器:基于PLM衍生的嵌入使用随机森林进行药物-疾病关联预测,提供鲁棒的预测结果。
-
Youden指数优化阈值选择:通过Youden指数确定最佳阈值,将概率分数转换为二进制预测。

记录AI蛋白质设计在诺奖背后的人和事
