 夜雨聆风
夜雨聆风





Day1——GEO及TCGA数据下载(以不变应万变)
首先,GEO数据库的下载不止一种方法,比如 GEoquery 包中的 getGEO 函数。
但是每种方法有每种方法的优缺点,比如 getGEO 下载好的文件比较易读,但是有的时候在R里下载可能出现各种小bug(比如链接超时、下载文件是空的…..),反正我刚开始学习的时候是十分茫然的。
今天给大家带来的,就是通过GEO官网搜索GES编号直接下载,步骤可能相较其他的繁琐一些,而且下载后的文件还需要自己处理一番,但是掌握后可以适用于所有GSE数据下载,并不容易出bug或出错(相当于以不变应万变了)
我们先找一下文章中所用了哪些上传到GEO的数据(文章中都会列出,直接在文献的页面上搜索 GSE 一般都能搜到)
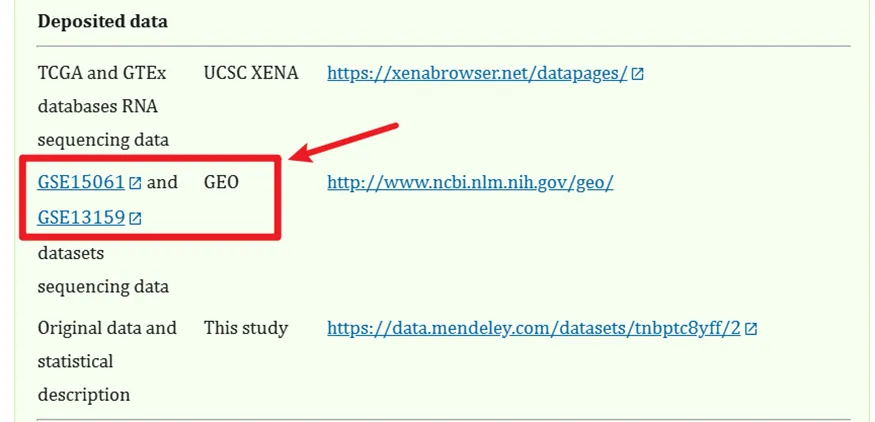
首先,我们进入GEO的官方页面,同时了解一下网站中的一些基本信息。
之前可以在浏览器直接搜GEO就可以找到,但现在直接搜出来的不是了,
没关系,我们搜索GSExxxxx(随便打几个数字),然后就能点进去了。
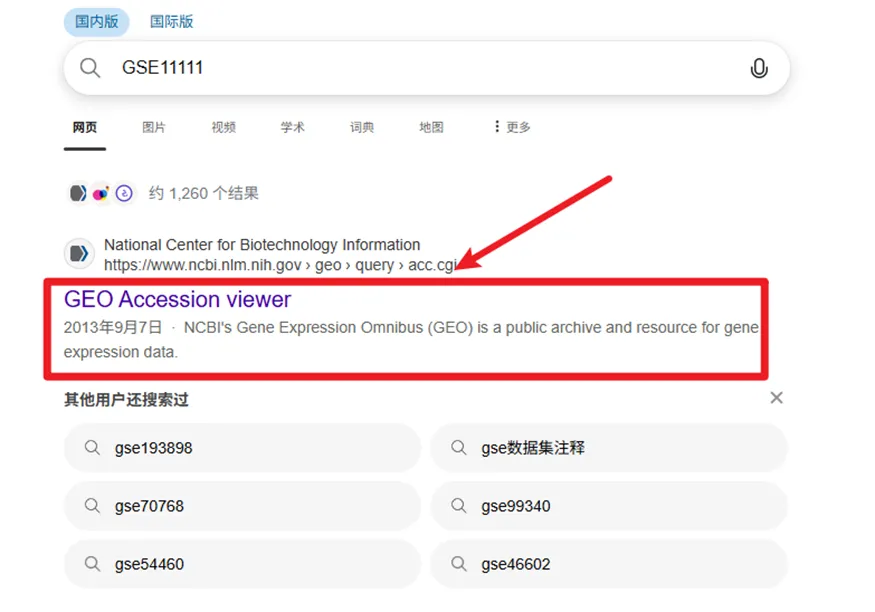
点进去后,在这里输入我们的GEO编号,然后点击GO即可
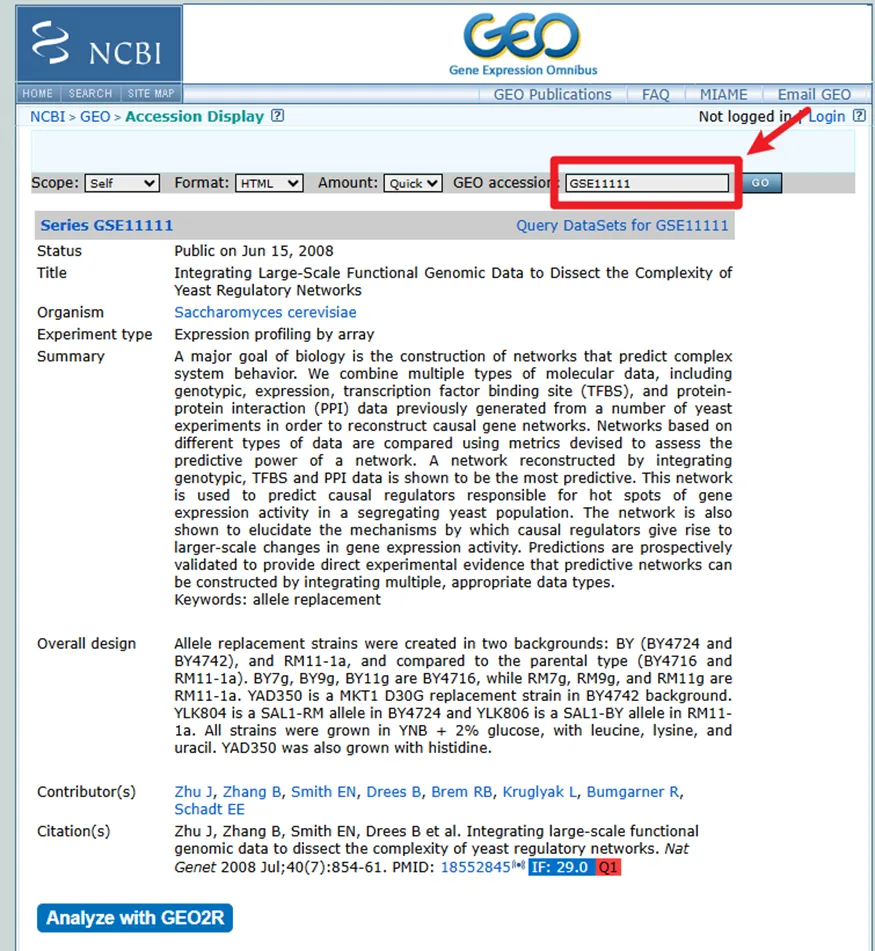
这里我们先以 GSE15061 为例,我们先看一下这个界面中的一些信息。

我们讲一下比较重要的4部分:
①数据类型,我们通常分析的是3类数据,
芯片—— array,
bulk转录组 或 单细胞转录组 —— high throughput sequencing
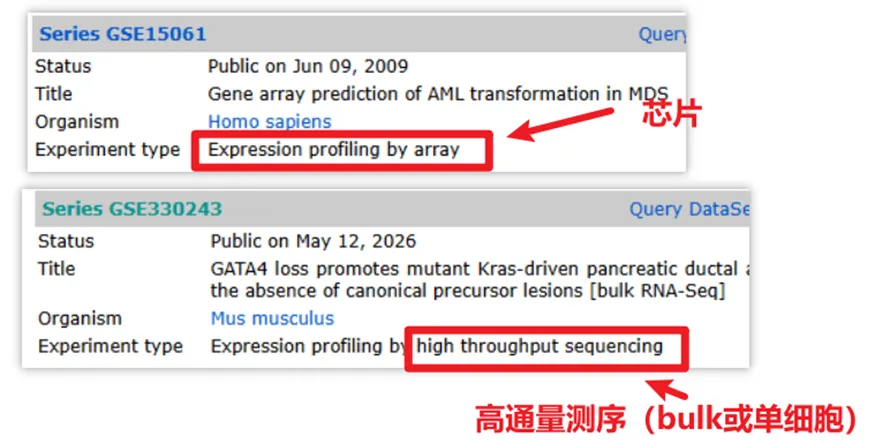
为什么这个重要呢?因为类型不一样其储存数据的地方也往往不同(后续会说明)。
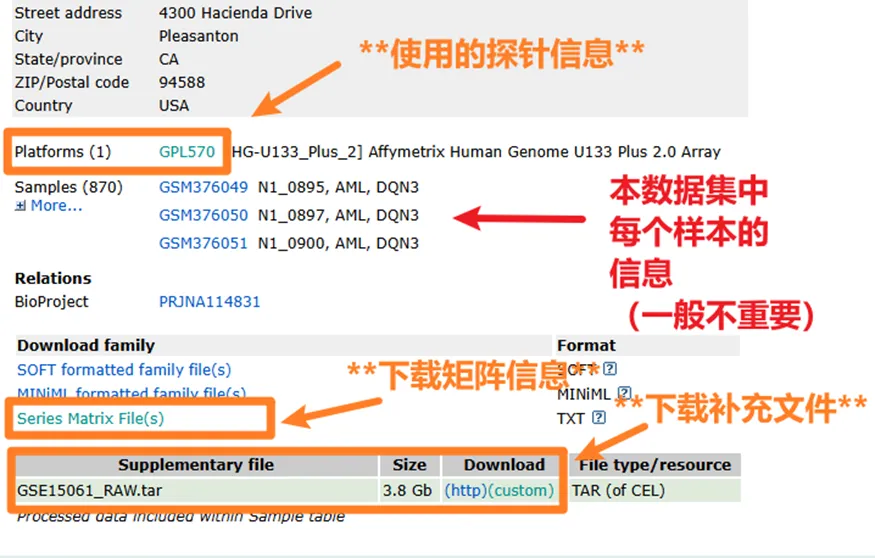
②探针信息(数据类型是array时需要注意)
探针是什么呢?简单来讲,芯片在检测的时候每个基因对应一个编号(如1007_s_at),我们需要将这些编号转变为我们认识的基因名称我们才能认识。
而使用不同芯片/探针去检测,有可能同一个编号对应不同的基因名称,所以可不能乱用。否则即便分析过程中不会报错,但是结果就可能是完全不正确的!!!

而当是高通量测序(单细胞或者转录组时),比较新的数据都是直接使用基因名做行名的,或者也是使用ENS编号指示基因名称的,一般使用 clusterProfiler 中的 bitr 函数就可以直接转换,就不需要太注重这个探针了。
③矩阵信息及补充材料:
这里讲放到一起进行说明,
如果数据类型是array(芯片)的话,临床信息跟表达矩阵往往都在Series Matrix Files里,如下。
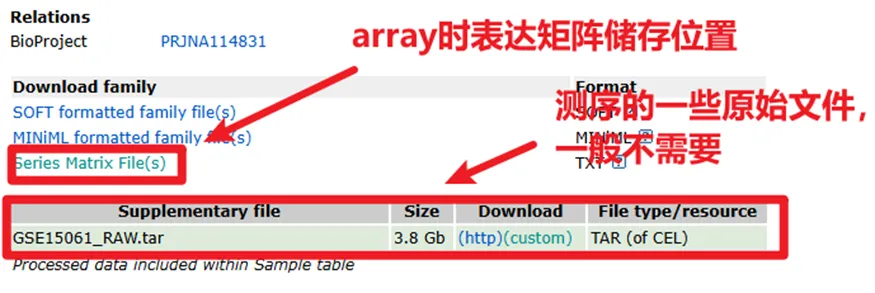
如果是high throughput sequencing(高通量测序,包括转录组+单细胞)的话,表达矩阵一般都在补充文件里。
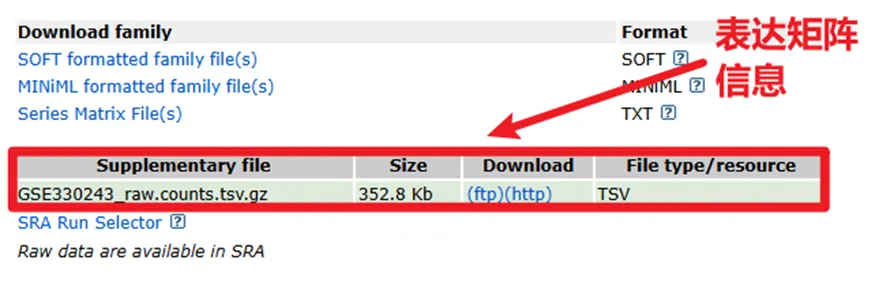
(当是high throughput sequencing时,Martrix File 里的文件也不会是空的,可能包含一些样本的基本信息等,我们按需下载即可)
经过上述的介绍,相信下载数据已经难不倒大家了,本次文章复现中使用了GSE15061和GSE13159这两个数据集,就按照下面的步骤一步一步下载下来即可。
(1)表达矩阵的下载:
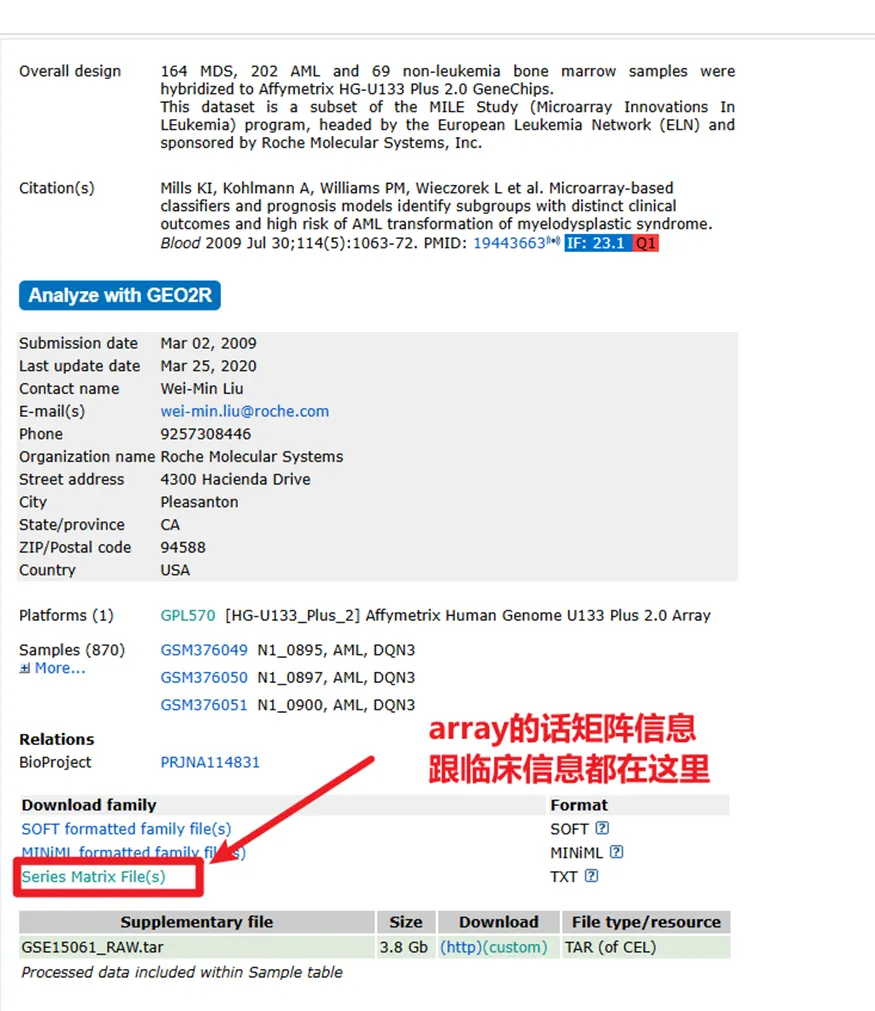
点进去

同样,我们找到GSE13159,也下载其表达矩阵与临床信息
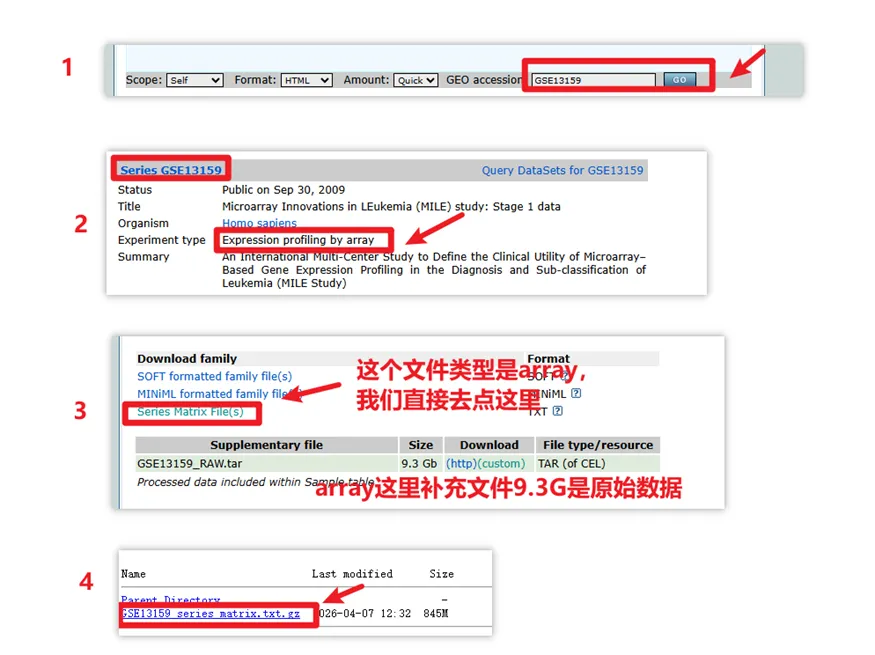
(2)注释文件的下载:
对于array文件,我们下载后的列名一般是像1007_s_at这样的探针名称,因此我们还要下载array对应的探针注释文件将其转换为基因。
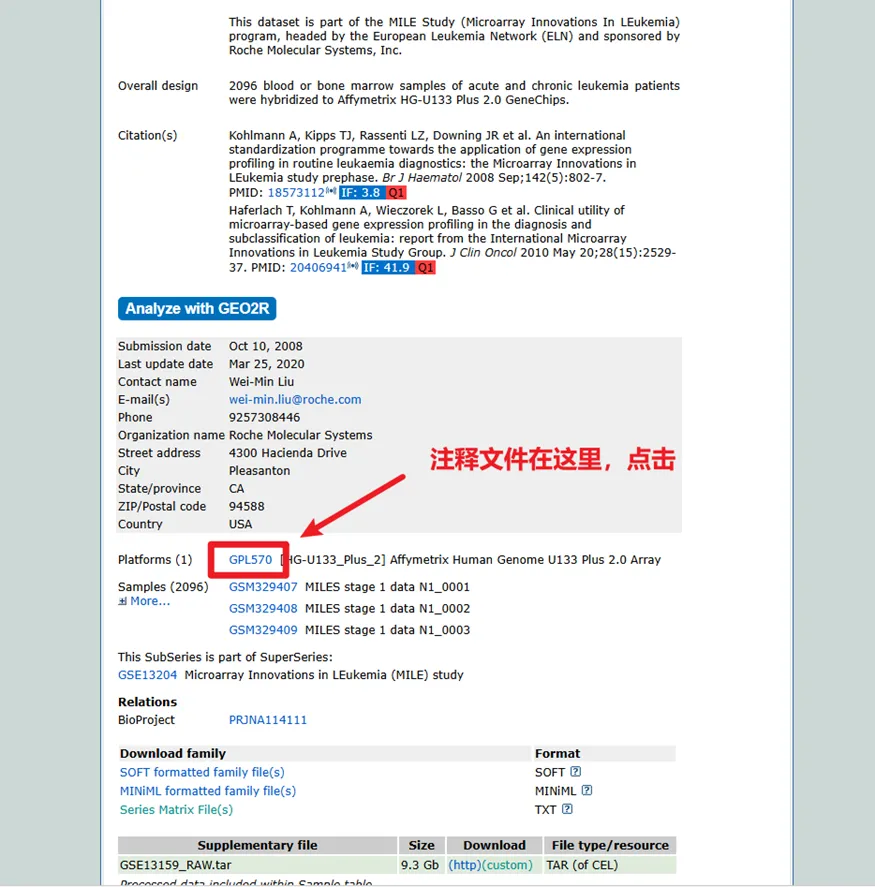
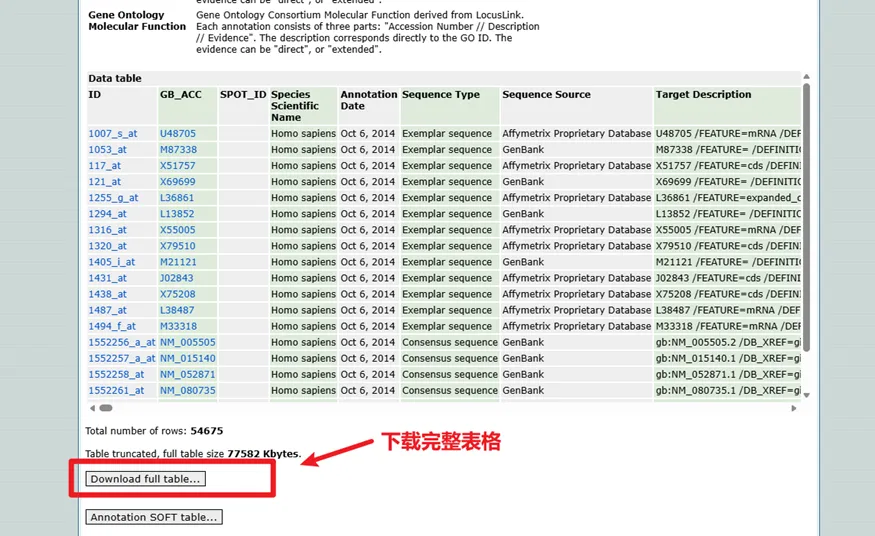
其中GPL570是最常见的array所用的探针,我们此次复现的文章中所使用的都是GPL570,因此我们下载一个即可。
下载完成之后,我们按如下方式整理好工作环境
(我还是喜欢用Rproj直接设置好工作环境,当然有的小伙伴习惯setwd也可以)
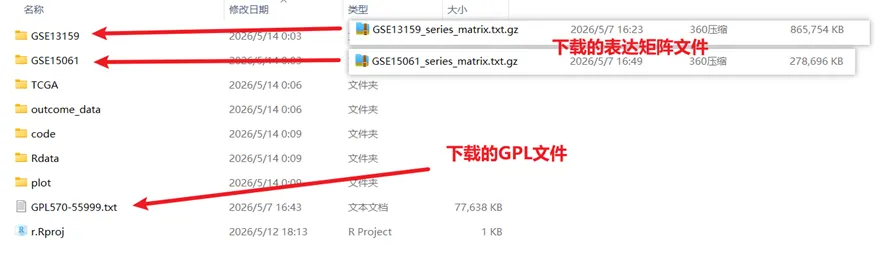
(其他的文件夹都是空文件夹,方便之后工作环境的整理)
–TCGA数据下载–
这里推荐从UCSC下载,对于初学者友好,整理比较完善
直接搜索
点进去后点Launch Xena
进到这个界面里点Data Sets
然后进入到这个页面,我们先把所有数据库的 √ 给勾掉,这样清爽一些。
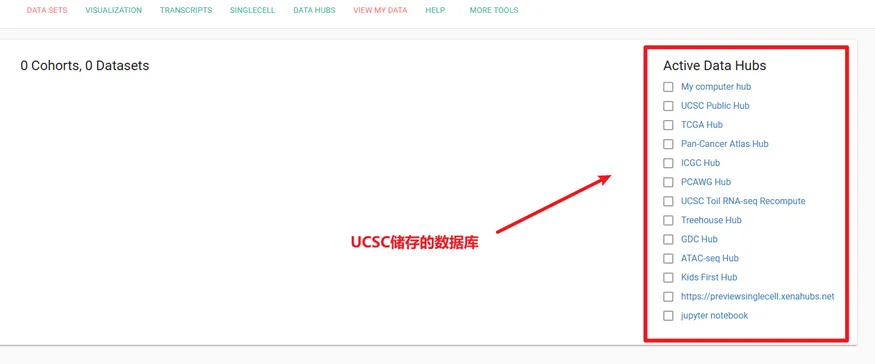
然后我们点什么呢?我们既然想要的是TCGA,那我们点 TCGA Hub 不就行了?
当然是没有什么问题的,但是更推荐的是,点 GDC Hub,
我们可以点进去大致看一眼。
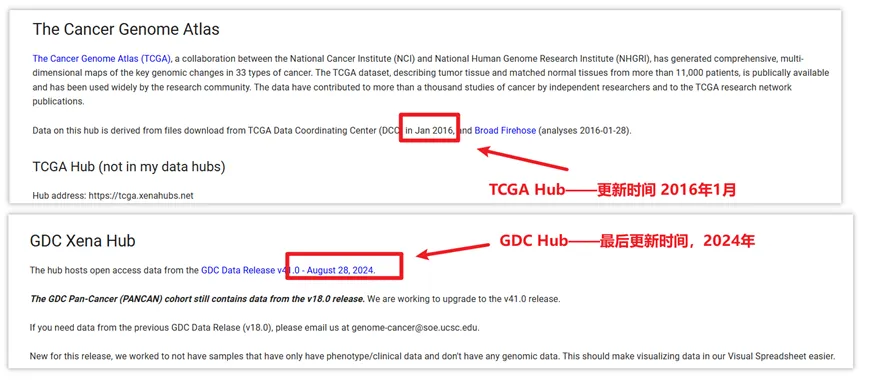
而且 GDC Hub 里是包含有TCGA数据的

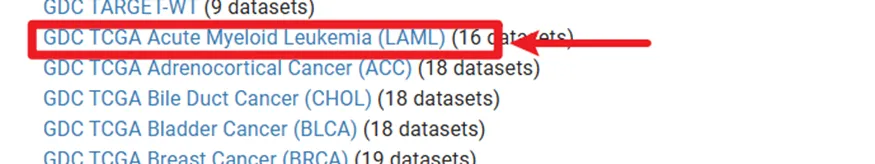
我们这里要的是急性髓系白血病,缩写是 LAML,不知道自己需要的癌症的缩写的可以直接百度搜索就知道了
点进去
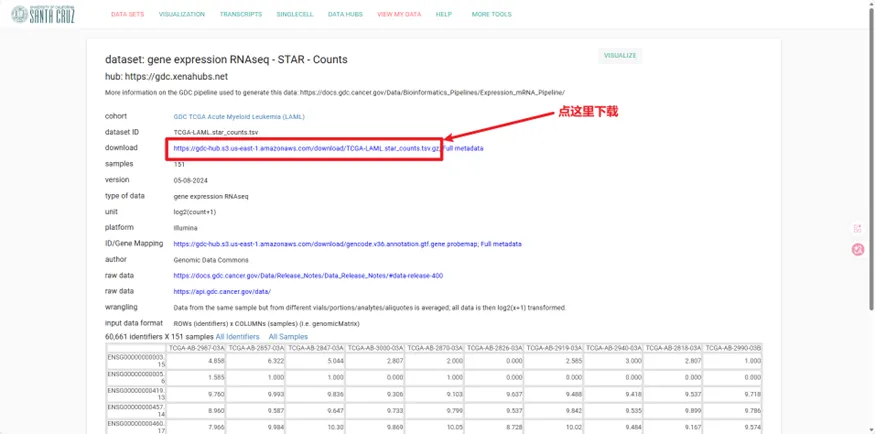
然后我们下载两个数据,一个是表达矩阵,一个是生存信息
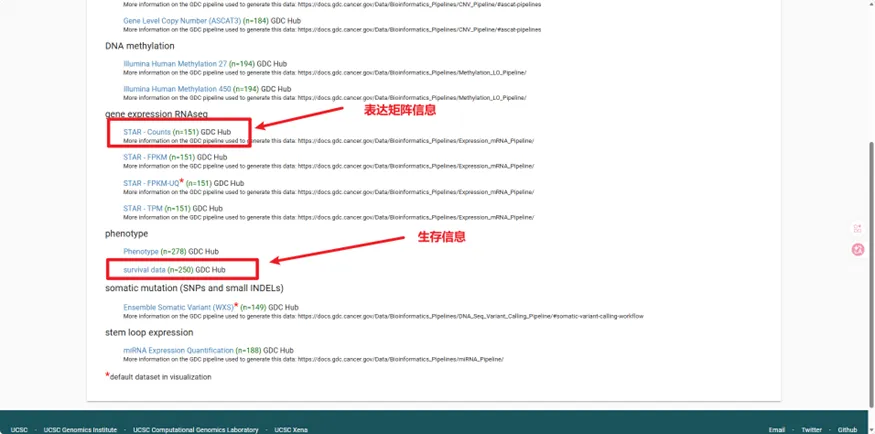
进去后点这里下载下
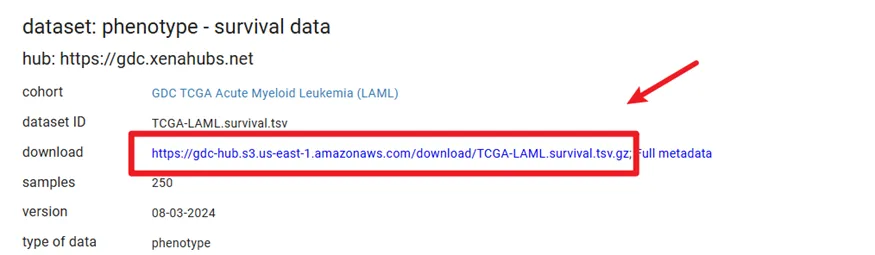
生存信息同理
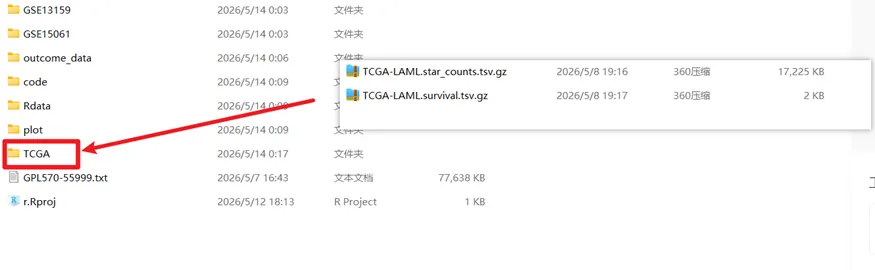
下载好的文件复制到我们工作环境下对应的文件夹即可。
明天的内容将讲述如何讲数据整理为分析前比较标准的格式。