 夜雨聆风
夜雨聆风【编者按】本文观点和数据均来自于开源信息,旨在促进专业讨论,不代表任何机构立场。其准确性、完整性,请读者自行甄别,并欢迎进一步研讨和交流。 |
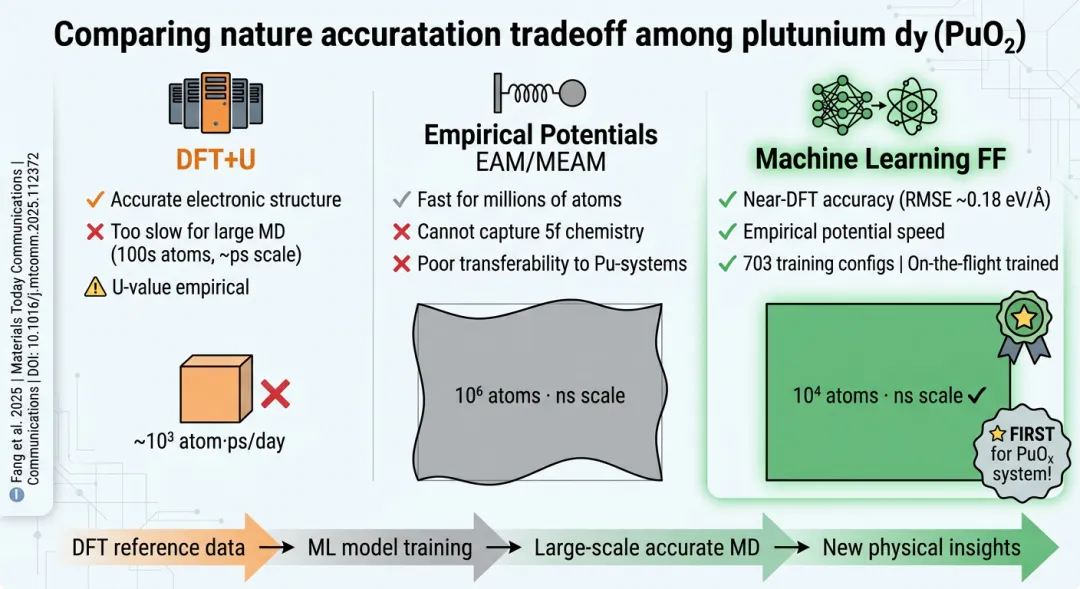
二氧化钚(PuO₂)是钚材料科学中最重要也最棘手的化合物之一——它是核燃料循环的核心物质、核废料长期储存的主要形态,也是钚金属表面腐蚀的终极产物。然而,这种看似简单的萤石结构氧化物背后隐藏着令第一性原理计算方法头疼了二十余年的电子结构难题。2025年4月,一项发表于《Materials Today Communications》的研究首次为PuO₂系统构建了专用机器学习力场(MLFF),标志着锕系氧化物的原子尺度模拟进入了一个全新阶段。

▌为什么PuO₂这么难算?
(一)5f电子的魔咒
要理解机器学习力场对PuO₂研究的意义,首先需要理解为什么传统方法在这类材料上举步维艰。
钚的原子序数为94,其价电子组态包含5f轨道电子。与3d过渡金属的d电子或4f系镧系元素的f电子不同,钚的5f电子处于一个极其微妙的关联能量区间:它们既不像轻元素的s/p电子那样完全离域化(可以用标准的密度泛函理论DFT很好地描述),也不像重镧系的4f电子那样高度局域化(几乎不参与成键)。5f电子的波函数在空间上适度扩展,能够参与化学键合;但同时电子间的库仑排斥作用又足够强,导致显著的电子关联效应(strong electron correlation)。这种"既不离域也不局域"的中间状态使得单一近似框架难以准确描述。
具体到PuO₂,这一困难集中体现在以下几个方面:
带隙性质的争议。PuO₂是一个绝缘体——这是实验确定的事实。但其本质是莫特-哈伯德绝缘体(Mott-Hubbard insulator,由在位库仑排斥驱动的关联绝缘体)还是电荷转移绝缘体(charge-transfer insulator,由电荷从配体向金属转移驱动的绝缘体)?实验测量的光学带隙范围跨度极大:从约1.8 eV到高达4.1 eV,不同测量技术给出的数值差异悬殊。标准DFT计算(即使使用杂化泛函如HSE06或PBE0)往往严重低估甚至无法打开带隙——这是DFT处理强关联系统的经典失败模式之一。
磁基态的不确定性。实验已明确证实PuO₂在宽温区(4–1000 K)内表现为非磁性(non-magnetic, NM)基态。但大量未经校正的DFT+U计算却错误地预测其为反铁磁性(AFM)基态。问题的根源在于:钚的5f壳层有7个轨道可填充4个电子,可能的电子占据方式多达35种,其中多种构型对应的总能量差异极小(小于0.2 eV/公式单元)。标准自洽计算容易收敛到某个低能激发态而非真正的基态——这就像在一个起伏极小的复杂地形中寻找最低点,算法很容易被某个看起来像谷底实际只是浅坑的位置所"欺骗"。直到Chen和Kaltsoyannis在2022年引入占据矩阵控制(Occupation Matrix Control, OMC)方法,对PuO₂进行系统性构型扫描后,才最终确认NM基态的正确性并推荐了合理的U值参数(U = 4.5 eV用于NM态),同时指出AFM态的能量仅比NM高不到0.18 eV/f.u.——如此接近的能量简并进一步凸显了计算的敏感性。
自旋轨道耦合(SOC)不可忽略。对于5f元素,SOC强度可达数个电子伏特量级,必须作为相对论效应纳入考量。这意味着即使是DFT+U级别的计算也需要额外叠加SOC修正,进一步推高了单点能量计算的代价。
(二)DFT+U的困境与DMFT的代价
面对上述挑战,计算材料学界的应对策略大致分为两个层级:
第一层级是DFT+U方法——在标准DFT哈密顿量中对5f轨道施加一个唯象的在位库仑排斥修正项(Hubbard U参数)。这种方法实现简单、计算效率适中,是目前研究锕系材料最广泛使用的工具。但其根本性局限在于:U值的选取缺乏严格的第一性原理确定方案,通常依赖于经验调参以匹配特定实验可观测量(晶格常数、带隙或形成能等);而不同的可观测量往往要求不一致的最优U值——用晶格常数优化得到的U可能给出偏差较大的带隙,反之亦然。更关键的是,DFT+U本质上仍然是一种静态平均场方法,它无法正确描述有限温度下的电子涨落动力学行为。
第二层级是DFT+DMFT(密度泛函理论结合动力学平均场理论)。DMFT通过将晶格模型映射到有效的杂质问题并精确求解(通常采用连续时间量子蒙特卡洛CT-QMC方法),能够在原则上完整处理动态关联效应。DMFT确实能给出比DFT+U准确得多的电子谱函数和热力学性质。但其计算代价极为高昂:单个温度点的自洽计算可能需要数天至数周的CPU时间,这使得基于DMFT的大规模分子动力学模拟在实际层面不可行——而分子动力学恰恰是研究缺陷扩散、界面演化、辐射损伤级联等过程动力学行为不可或缺的工具。
于是我们面临着一个尴尬的方法论鸿沟:能够提供足够精度来描述PuO₂电子结构的计算方法太慢而跑不动分子动力学;能跑动力学的经验力场又太粗糙而无法捕捉复杂的化学键合环境依赖性。 这个鸿沟就是机器学习力场要填补的空间。

▌机器学习力场:跨越精度的桥梁
(一)核心思想
机器学习力场的概念本身并不复杂。其核心思想可以概括为一句话:用一个回归模型(通常是神经网络或核方法)去拟合由高精度第一性原理计算给出的原子势能面(Potential Energy Surface, PES)。一旦训练完成,这个ML模型就可以在几乎不损失精度(相对于参考DFT方法)的前提下,以快几个数量级的速度预测任意原子构型的能量和原子受力——从而使得以前因计算成本过高而无法开展的大规模原子模拟变为可能。
与传统经验势函数(如EAM、MEAM等)相比,MLIP的关键区别在于:经验势函数使用固定的解析形式(预先设定好的数学函数),其中的参数数量有限(通常几十到几百个),拟合过程本质上是用简单函数去逼近复杂PES;而MLIP使用的是高度灵活的非参数化模型,具有数以万计乃至百万计的可调参数,理论上可以任意精度地逼近任何连续PES(通用逼近定理保证了这一点)。当然,灵活性的代价是需要更大规模的高质量训练数据集和更谨慎的过拟合控制策略。
(二)主流技术路线
当前MLIP领域存在几条主要的技术路线:
高斯近似势(GAP - Gaussian Approximation Potential)。由剑桥大学Gábor Csányi团队开发,使用高斯过程回归作为核心模型,以原子簇的多体展开描述符(如SOAP - Smooth Overlap of Atomic Positions)表征局部结构环境。GAP在精度和可解释性方面表现优异,但预测阶段的计算开销随训练集大小增长,在大体系上的效率受到一定限制。
神经网络势(Neural Network Potential, NNP)。以Behler-Parrinello型对称函数为原子描述符,以前馈神经网络为回归模型。DeepMD(深度势分子动力学)是该路线近年来的突出代表,通过引入等变性和注意力机制等新设计大幅提升了效率和表达能力。NNP在预测速度上具有优势,适合大规模并行计算。
谱邻域分析势(SNAP - Spectral Neighbor Analysis Potential)。由Sandia国家实验室开发,使用广义球谐函数展开的邻域密度谱系数作为描述符,线性回归拟合。SNAP的优势在于简洁性和良好的外推行为。
VASP内置的On-the-fly MLFF。这是Fang等人采用的方案。该方法直接嵌入在VASP程序包中,在从头算分子动力学(AIMD)运行过程中自动监测DFT计算的力和能量的预测误差,当误差超过阈值时自动将当前构型加入训练集并在线更新ML模型。整个流程实现了从数据生成到模型训练的全自动化,无需手动构建和管理训练集。

▌PuO₂的第一个机器学习势函数
(一)论文概况
论文题为《First-principles study of the properties of plutonium oxides and their interfaces based on machine learning》(基于机器学习的钚氧化物及其界面性质的第一性原理研究),发表于2025年4月的《Materials Today Communications》(第45卷,文章编号112372),第一作者Fang Yilin,通讯作者Wang Jintao,DOI: 10.1016/j.mtcomm.2025.112372。https://www.sciencedirect.com/science/article/pii/S2352492825008840
(二)技术方案详解
DFT参考方法的选取。Fang等人选择以DFT+U(文中记作MLFF_U)作为生成训练数据的基准第一性原理方法,计算平台为VASP。这一选择的合理性在于:虽然DFT+U并非最精确的电子结构方法(DMFT才是),但它提供了精度与效率之间的最佳平衡——足以给出可靠的基态结构、弹性性质和缺陷能量信息,同时又能在合理时间内产生数百个高质量训练构型所需的DFT参考计算。考虑到后续MLFF的目标应用场景(分子动力学模拟中的能量和力预测),DFT+U级别的参考精度已经完全满足需求。
On-the-fly训练流程。该工作采用了VASP内置的即时机器学习方法来构建MLFF。整个流程在AIMD模拟过程中自动执行:首先从一个初始的小型结构数据库出发开始MLFF辅助的分子动力学模拟;在每一步AIMD中,如果MLFF预测的力和能量与实时DFT计算结果的偏差超过预设阈值(通常力的RMSE阈值约为0.1–0.2 eV/Å量级),则将该构型自动添加到训练数据集中并更新模型;随着模拟的推进,数据集不断扩充、模型持续优化,MLFF的预测精度逐步提升直至达到稳定状态。这种方法的优势在于:训练数据的采样空间自然覆盖了AIMD轨迹访问过的相空间区域,避免了手动构建数据集时可能出现的关键构型遗漏。
训练集构成。最终生成的数据集共包含703个独立的结构配置,按体系分类为:PuO₂配置270个、Pu₂O₃配置123个、其他(主要为PuO₂/Pu₂O₃界面及相关结构)310个。700余个训练构型对于二元氧化物体系的MLIP而言属于中等偏小的规模——这得益于on-the-flow方法高效的数据利用率:每个构型都来自于实际的动力学演化路径,信息含量高且冗余度低。
验证精度。报告的主要验证指标包括:能量和力的均方根误差(RMSE)分别达到0.0405 eV/Å和0.1807 eV/Å(原文单位引用如此)。作为参照,一般认为力的RMSE低于0.15–0.20 eV/Å即可认为MLFF达到了可用于生产级别分子动力学模拟的精度要求。此外,研究团队还通过计算弹性常数和声子色散关系对力场进行了交叉验证——这些派生性质的计算结果与此前的DFT研究高度吻合,进一步确认了MLFF的可靠性。
(三)科学发现:界面演化的原子图景
除了方法学层面的贡献外,Fang等人的工作还利用训练好的MLFF对PuO₂/Pu₂O₃界面系统进行了案例研究,揭示了一个重要的物理机制:
在PuO₂与Pu₂O₃的界面处,氧原子的扩散驱动了结构重构过程。具体来说,界面区域的氧离子在热激活下发生迁移,使整个界面体系从初始的高能亚稳构型逐渐弛豫到更低能量的稳定排布。重构后的界面能降低至5.72 J/m²——这是一个相当低的界面能值,意味着PuO₂/Pu₂O₃异质界面的形成在热力学上是容易进行的。
声子分析提供了补充证据:重构后的振动频率谱受到明显抑制(即声子频率整体降低),这与结构稳定性增强的图像一致——更深的热力学阱对应着更高的结构刚性。
这一发现的实际意义在于:PuO₂/Pu₂O₃界面在钚材料的腐蚀演化过程中可能扮演着关键角色。当金属钚暴露于含氧环境中时,表面首先氧化生成PuO₂层;随着氧化程度加深或局部氧化学势升高,部分区域可能进一步转化为低价氧化物Pu₂O₃。这两种氧化物之间的界面性质直接影响氧化层的保护性行为和离子传输特性——而这正是理解钚长期腐蚀动力学的微观基础。

▌对核材料研究的深远影响
(一)填补模拟能力的空白
Fang等人开发的PuO₂ MLFF填补了一个此前长期存在的模拟能力空白。在此之前,研究者若要对含PuO₂体系进行大规模原子模拟,只能在以下选项中做出妥协:使用DFT直接做AIMD——但受限于计算成本只能模拟几百个原子在几十皮秒时间尺度内的行为;或者使用为UO₂开发的经验势函数(如Morelon势、Basak势等)勉强套用于PuO₂——但由于Pu 5f与U 5f的电子结构差异显著,这类套用的可靠性难以保证。
有了专用的MLFF之后,包含数千甚至数万个原子的PuO₂体系在纳秒时间尺度的分子动力学模拟首次变得可行。这意味着研究者现在可以系统地考察以下此前无法触及的问题:氦气泡在PuO₂基质中的成核与生长动力学、辐照产生的点缺陷(氧空位、Pu空位、Frenkel缺陷对)的长程扩散和聚集行为、多晶PuO₂中沿晶界的优先输运通道、以及PuO₂/Pu₂O₃/Pu多层界面体系的协同演化机制等。
(二)与钚老化问题的连接
回到本系列的中心主题——钚弹芯老化。PuO₂ MLFF的发展与老化研究之间存在着多重联系:
氦在PuO₂中的扩散。α衰变产生的氦不仅滞留在金属钚中形成氦泡,也会在表面PuO₂氧化层中累积。理解氦在PuO₂晶格中的扩散行为(扩散系数、扩散路径、活化能、以及与缺陷的耦合作用)对于评估氧化层的完整性破坏风险至关重要。MLFF使大规模的氦扩散分子动力学模拟成为可能。
辐射损伤的长期演化。α反冲核在穿过PuO₂层时会留下位移损伤级联。损伤后的缺陷如何随时间演化?是否会发生非晶化转变?缺陷复合的动力学速率是多少?这些问题都需要长时间尺度的模拟才能回答。
氧化层的保护性退化。PuO₂作为钚金属表面的钝化层,其保护性能是否会因自辐照损伤积累和氦泡形成而随时间退化?这是一个涉及多物理场耦合(辐照+扩散+应力)的复杂问题,MLIP为多尺度模拟链提供了关键的原子级输入数据。
(三)未来方向与挑战
尽管Fang等人的工作具有开创性意义,但PuO₂ MLIP领域仍处于起步阶段,若干重要的挑战有待解决:
训练集的完备性问题。703个构型的训练集对于体相性质和简单界面的描述已经足够,但要涵盖辐照损伤级联产生的高度无序结构、高温液相或非晶相等极端状态,数据集还需要大幅扩充。特别是涉及大量点缺陷和高浓度氦的构型——这些恰好是老化研究最关心的场景——目前可能尚未被充分采样。
外推可靠性的边界。所有机器学习模型的通病是对训练分布之外的数据外推能力差。MLFF在远离训练数据覆盖的区域(如极高的压力、从未访问过的新缺陷构型等)可能给出完全不可靠的预测而不发出任何警告。建立针对锕系MLIP的外推检测和不确定性量化方法是亟待发展的配套技术。
从PuO₂到更多锕系化合物。钚的氧化物不止PuO₂和Pu₂O₃两种——还包括多种非化学计量比的氧化物相(PuO₂₊ₓ,其中x可在0到约0.25之间变化)以及更高价的氧化物。每种化合物都有其独特的电子结构和化学行为,需要发展各自的专用MLFF或构建统一的锕系氧化物通用力场。
与实验数据的闭环校验。任何计算工具的最终价值都需要通过实验验证来确认。PuO₂ MLFF预测的各种性质(扩散系数、缺陷形成能、界面反应速率等)需要与可获得的实验数据进行系统比对——这将是一个需要计算学家和实验学家紧密合作的漫长过程。

▌结语
Fang等人2025年的工作标志着锕系材料模拟进入了一个新的方法论阶段:从"要么够准但太慢要么太快但不准"的两难选择,走向了"既够准又够快"的第三条道路。机器学习力场不是要替代第一性原理计算——相反,它是以DFT为根基、以数据为桥梁、以机器学习为引擎的新型多层次模拟范式的关键组件。
对于从事金属铀/钚表面氧化腐蚀多尺度理论计算研究的科研人员而言,这项工作的意义尤为直接:它提供了一个现成的、经过验证的工具起点,可以在此基础上进一步发展适用于自己研究体系的专用力场——无论是纯金属铀/钚、铀/钚氧化物、还是更复杂的合金-氧化物界面系统。在这个意义上,PuO₂的第一个AI势函数不仅是一项独立的科研成果,更是整个锕系计算材料学领域迈向智能化、高通量化的一个信号枪。


