 夜雨聆风
夜雨聆风
在当今的数字医疗浪潮中,开发“作为医疗器械的软件”(Software as a Medical Device,简称SaMD)无疑是最具潜力的赛道之一。然而,对于无数医疗科技初创公司和转型中的传统械企而言,在产品正式推向市场之前,往往会迎头撞上第一座大山——产品分类(Classification)。
您的软件属于什么风险等级?这不仅仅是一个学术问题,它直接决定了您的监管路径、文档要求,并在很大程度上决定了产品上市的时间表和成本预算。
今天,我们将为您系统性地拆解不同地区的 SaMD 分类标准及核心合规要求,帮您拨开监管迷雾,为产品的全球化战略打下坚实基础。
01
美国 FDA 体系:
从 SaMD 到 DSF 的演进与风险分级
在美国,由于“作为医疗器械的软件”本质上仍是医疗器械,FDA 对 SaMD 的分类逻辑与传统实体医疗器械保持一致:基于风险程度,将其划分为I 类(Class I)、II 类(Class II)和 III 类(Class III)。
I 类(Class I)
风险最低,通常只需满足一般控制(General Controls),如企业注册、产品列名、遵守质量体系要求等,多数可豁免上市前通知。
II 类(Class II)
具有中等风险,通常需要特殊控制,并要求提交510(k) 上市前通知,证明其与已上市的“实质等同”产品(Predicate Device)具有一样的安全性和有效性。
III 类(Class III)
风险最高(如维持生命、植入体内或存在重大不合理风险),必须通过极为严格的上市前批准(PMA)途径,提供详实的临床数据。
监管视角的微调:从 SaMD 到 DSF
值得注意的是,在 FDA 近期发布的一系列指南文件中,官方开始越来越多地使用“器械软件功能”(Device Software Function, DSF)这一术语,来替代业内熟知的 SaMD。
根据《联邦食品、药品和化妆品法案》(FD&C Act),DSF 被定义为任何符合医疗器械定义的软件功能。这种术语的转变主要是为了统一监管口径,因为很多时候软件既可以独立存在(SaMD),也可以嵌入在硬件中(SiMD)。虽然名称变了,但底层的监管义务并没有实质性改变。
文档提交要求:基础型 vs. 增强型
在上市前提交的文档要求上,SaMD(或 DSF)与传统硬件设备有着显著的区别。FDA 在关于《器械软件功能上市前提交内容》的最新指南中,彻底废除了过去“次要、中等、重大”的关注级别(Level of Concern),取而代之的是两个全新的文档等级:增强型(Enhanced)与基础型(Basic)。
增强型文档(Enhanced Documentation)
如果设备软件功能的故障或缺陷可能导致出现危险情况,并且在采取任何风险控制措施之前,评估出存在导致患者死亡或重伤的可能风险,则必须提交增强型文档。
基础型文档(Basic Documentation)
适用于所有不符合增强型标准的其他情况。
核心避坑指南:文档级别 ≠ 风险等级
很多研发人员会误以为文档级别与 FDA 风险等级是一一对应的。实际上,它们完全是独立评估的!
一个 Class III 的高风险设备几乎肯定需要增强型文档;但是,一个 Class I 或 Class II 的设备,如果其某项软件功能在特定极端情况下可能导致严重后果,同样也需要提交增强型文档。制造商必须为每一款设备进行独立的深入评估。

02
FDA 的特殊豁免:
您的软件究竟算不算 CDS?
在谈论医疗软件时,还有一个绝对绕不开的产品类别:临床决策支持(Clinical Decision Support, CDS)软件。这类产品处于监管的灰色地带——它到底算不算受 FDA 监管的医疗器械,完全取决于它是否满足特定的法定标准。
《21世纪治愈法案》(21st Century Cures Act)对 FD&C Act 进行了修订,增加了第520(o) 条。该条款明确规定,如果某些软件功能同时满足四项特定标准,就可以被排除在医疗器械的定义之外,从而免受 FDA 监管。
根据 FDA 发布的 CDS 最终指南,要成为豁免的“非器械CDS(Non-Device CDS)”,软件功能必须同时满足以下全部四个条件(缺一不可):
1
非信号/图像处理
软件不得获取、处理或分析来自诊断设备的医学图像或信号(如 ECG 信号、X 光片、生理监测波形等)。
2
展示医学信息
软件的功能是显示、分析或打印有关患者的医学信息或基于文献的医学知识。
3
提供辅助建议
软件旨在为医疗保健专业人员(HCP)提供有关疾病预防、诊断或治疗的支持或建议。
4
独立审查依据
软件必须使该专业人员能够独立审查这些建议的依据,而不是强迫他们将这些建议作为临床决策的主要或唯一驱动力。
为什么第 4 点是“AI 杀手”?
如果软件功能满足上述所有四个标准,它就被视为“非器械 CDS”,不在 FDA 监管范围内。但如果未能满足其中任何一项,它就仍然受监管的医疗器械软件。
对于目前的 SaMD 制造商来说,这既是机遇也是挑战。许多基于深度学习和人工智能(AI/ML)的产品往往折戟于第 4 项标准。因为深度学习模型往往是“黑盒(Black Box)”,医生无法知晓算法是如何得出某个诊断建议的(即无法“独立审查依据”)。因此,大多数现代 AI 诊断辅助软件都被 FDA 归类为受监管的医疗器械。
如果您正在开发旨在支持临床决策的软件,及早根据这四项标准进行严格评估,可以帮助您准确界定产品属性,避免在准备 510(k) 申报时才突然发现自己走错了路线。
延伸推荐
如您持续关注中国医疗器械出海美国,欢迎关注Protheragen 服务号,获取更多 FDA 合规干货和实操指南。
点击 Protheragen

03
欧洲 EU MDR 与 Rule 11
将视线转向欧洲。
在欧盟,SaMD 受欧盟医疗器械法规(EU MDR)或体外诊断医疗器械法规(EU IVDR)的监管。欧盟不使用 SaMD 这个词,而是统称为“医疗器械软件”(Medical Device Software, MDSW)。
其风险分类系统与传统设备相似:分为Class I、Class IIa、Class IIb 和 Class III。
使欧盟软件分类系统独树一帜、也让无数企业头疼的,是欧盟 MDR 附件 VIII 中著名的规则 11(Rule 11)。Rule 11 专门为医疗器械软件提供了具体的分类逻辑:
默认 Class IIa
旨在提供用于诊断或治疗决策信息的软件,默认分类为 Class IIa。
升级至 Class III
如果上述决策可能导致患者死亡或不可逆转的健康恶化,则直接升级为最高风险的 Class III。
升级至 Class IIb
如果决策可能导致严重的健康恶化或需要进行外科手术干预,则归为 Class IIb。
生理监测软件
旨在监测生理过程的软件通常为 Class IIa;但如果它监测的是关键生命体征,且这些参数的危险性变化可能对患者造成直接危险(如 ICU 里的实时心电监控),则升级为 Class IIb。
Class I
所有不符合上述任何升级条件的“其他”软件才属于 Class I。
告别“轻松的 Class I”时代
在旧的 MDD 指令时代,许多医疗软件可以通过自我声明轻松归为 Class I。
但在 MDR Rule 11 的框架下,绝大多数提供诊断或治疗信息的医疗器械软件,起步就是 Class IIa。这意味着它们必须经过欧盟公告机构(Notified Body)的严格审查才能获得 CE 标志。
对于计划进入欧洲市场的制造商而言,欧盟委员会发布的MDCG 2019-11和 MDCG 2021-24行业指南是必读的圣经,其中详细解释了如何应用 Rule 11 的各种边界情况。
04
IMDRF 分类框架:
全球监管的“通用语言”
国际医疗器械监管机构论坛(IMDRF)也开发了一套独立的软件风险分类框架。虽然 IMDRF 本身不是一个具有执法权的监管机构,但它的成员包括了全球最大的医疗器械市场代表(美国、欧洲、加拿大、澳大利亚、日本、中国、巴西等)。IMDRF 致力于加速国际监管趋同,其工作组对全球法规的走向具有深远的指导意义。
IMDRF 的 SaMD 分类系统采用二维矩阵模型,分为四个类别(Category I 到 IV):
纵轴(信息的重要性)
该 SaMD 提供的信息对医疗决策的干预程度有多深?
分为三个层次:
用于治疗或诊断(Treat or diagnose)
驱动临床管理(Drive clinical management)
为临床管理提供参考信息(Inform clinical management)
横轴(医疗状况的严重性)
该软件旨在应对的疾病或患者状况有多严重?
分为三个层次:
危急(Critical)
严重(Serious)
非严重(Non-serious)
结合这两个维度,可以矩阵化地将软件分为 Category I(风险最低,如非严重状况的参考信息)到Category IV(风险最高,如危急状况下的治疗/诊断决策)。
通往欧盟合规的桥梁
IMDRF 分类通常不作为单一国家的直接法定分类系统。但它的巨大价值体现在欧盟合规上——前文提到的欧盟官方指南 MDCG 2019-11 中,专门包含了一张将 IMDRF 类别映射到欧盟 MDR 风险等级的对照表。
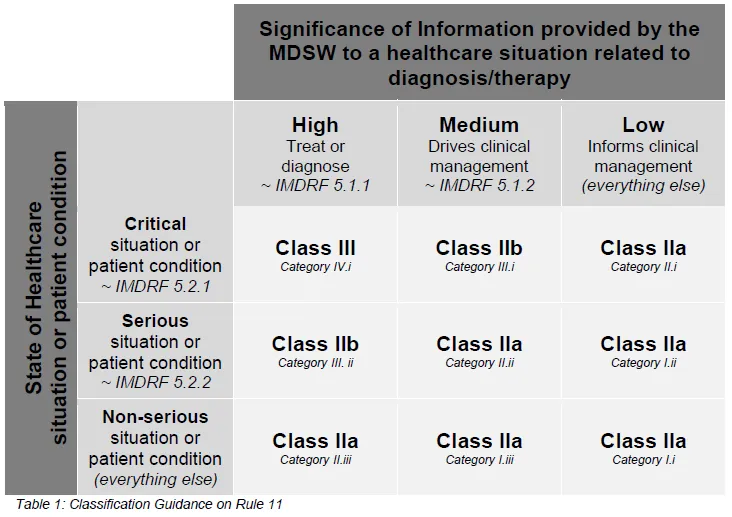
图源:health.ec.europa.eu
如果您是第一次面对晦涩难懂的 Rule 11,强烈建议您先通过 IMDRF 框架理清您软件的业务逻辑和风险底线,再将结果代入欧盟的特定标准中,这会极大理顺您的分类思路。
05
IEC 62304 软件安全级别:
研发团队的“紧箍咒”
跳出纯粹的行政法规监管圈,我们必须谈谈工程师团队最熟悉的标准:IEC 62304《医疗器械软件 - 软件生命周期过程》。这不仅是一项国际标准,更是 FDA 认可的共识标准,遵循该标准被公认为满足软件安全合规的最佳实践。
IEC 62304 根据软件故障可能造成的伤害严重程度,设立了三个“软件安全级别(Software Safety Classification)”:
Class A
不可能造成任何伤害或健康损害。
Class B
可能造成伤害,但不是严重伤害。
Class C
可能造成死亡或严重伤害。
澄清误区:安全级别≠监管风险等级
准确理解 IEC 62304 的定位至关重要。IEC 62304 下的安全分类,并不是用来判断您的软件最终有多“安全”的,而是用来决定您的软件研发生命周期(SDLC)需要有多么严苛的。
分类越高,您需要执行的开发流程、架构设计、单元测试、集成测试、缺陷跟踪和验证活动就越繁杂。实际设备的安全性取决于您扎实的设计和开发过程,而不仅仅是一个分类标签。
千万记住,IEC 62304 的安全级别并不直接对应于 FDA 或欧盟的风险等级。虽然两者具有很强的相关性(例如,一个 Class C 的软件很可能在美国或欧盟都是Class III 高风险设备),但完全有可能出现这样的情况:
您的软件在IEC 62304 下因为某个潜在的严重故障点被定为 Class C,需要极其严谨的开发代码,但在 FDA 监管层面,通过综合风险控制后,它只被认定为Class II 设备。这两种判定是使用不同标准独立进行的。
点击图片查看对全球主要监管机构医疗器械法规的解读



扫码获取定制服务方案
我们是谁?
我们能为您做什么?
合规服务
法规解读、培训与咨询
产品准入路径规划
SaMD 产品分类评估
申报文档准备与提交支持
多监管框架差异分析
数字医疗全球准入

Proregulations 专注于数字医疗产品的全球合规策略,涵盖软件监管界定、安全级别辅导及多区域注册路径规划,助力企业构建清晰高效的医疗软件上市准入体系。如果您对我们的服务感兴趣,欢迎联系和咨询Proregulations(info@proregulations.com),我们将竭诚为您服务。

推荐阅读
指南解读:FDA 拟对 NIOSH 认证呼吸器(含医用 N95)实行大范围监管豁免
510(k)申报经验分享:没有强制国标,“自愿性标准”到底该怎么理解?
声明:本文内容由 Proregulations 整理,感谢关注。如需转载,请注明来自 Proregulations。
END

扫码关注
微信公众号:Proregulations
更多全球各行业的监管合规解决方案
网址:https://www.proregulations.com
邮箱:info@proregulations.com
