 夜雨聆风
夜雨聆风先导化合物优化是药物发现的重要阶段,从苗头化合物(hit)到临床候选药物(candidate),研究人员需要对分子进行数十至数百轮迭代的结构修饰,以同时优化活性、选择性、ADMET性质和合成可及性。分子编辑的兴起为这一阶段带来了效率的提升 [1,2],其在已有分子骨架上进行原子尺度的精准"手术式"修饰,大幅简化结构改造的合成路径。与此同时,深度学习和强化学习在分子设计领域取得了显著进展[3-8],提供了一种系统性地探索化学空间并朝向多目标最优方向搜索的框架。
不过,把分子编辑嵌入计算框架有一个基础问题常常被 RL 算法设计和网络架构等下游讨论盖过去:分子编辑操作本身从哪来?
考察现有方法,答案明显地分化为两条路线。首先是规则驱动路线:将专业知识手工编码为操作规则,比如 MolDQN[9]用元素周期表和价键理论定义"哪些原子可以添加、哪些键可以形成";SHARP[11]用BRICS的16条化学键切断规则定义"分子在哪里可以被切割和替换";PGFS[25]用97条手工筛选和修复的反应模板定义"哪些化学转换是合法的"。这条路线的优势在于每条规则都有明确的化学依据,劣势在于规则的覆盖受专家知识瓶颈限制,比如 16条BRICS规则永远无法覆盖文献中所有已知的化学键切断模式。第二条是路线是基于数据驱动,即从大规模实验数据库中统计挖掘操作模式,其实也可以说是隐式的规则驱动。 ReACT-Drug[24]用mmpdb从ChEMBL自动推导数千条反应模板;MMPT-FM[29]从263万条匹配分子对中提取并去重得到80万条变换;CReM[30]通过统计哪些片段在相同化学上下文中出现过,来定义片段互换性。这条路线的优势在于自动覆盖长尾变换,80万条变换远超任何化学家团队能够手工枚举的数量,劣势在于变换的质量和覆盖度完全依赖训练数据的规模与质量。
这两条路线并非互斥,SHARP的SEM合成可行性掩码是在BRICS规则生成的片段上训练的(规则定义操作空间,数据训练约束模型),FREED[23]用CReM的数据驱动片段库搭配RL的策略网络。两条路线的边界在实践中是模糊的,但"操作知识从何而来"这一根本区分仍然具有分析价值,它决定了框架的可解释性、覆盖度和对新化学类型的适应能力。
严格来说,数据驱动也是一种规则驱动,只不过是隐式规则。 CReM 从 ChEMBL 统计出「在相同 3 键上下文中片段 A 和片段 B 可互换,频率 47」,这本身就是一条规则,只不过写规则的人从化学家换成了统计算法,规则的载体从 SMARTS 字符串换成了共现频次表。DigFrag 用 GNN 学「在哪里切断」,学出来的注意力权重就是隐式的断键规则。MMPT 的 80 万条变换,每条都是一条 if-then 规则。所以两条路线的对立不是「有规则 vs 没规则」,而是显式规则与隐式规则的区分。 显式规则由人写,语义可追溯到具体化学原理,可解释但覆盖有限;隐式规则由数据学,语义不可追溯,覆盖广但无法解释。遇到训练数据外的新化学类型,显式规则靠原理外推仍然成立,隐式规则因频率为零而无法生成。两条路线的对立本质上是规则编码方式的对立,以及由此带来的可解释性-覆盖度的区别。
常见分子编辑策略
骨架编辑与外围编辑
根据2024年J. Med. Chem.综述[1],分子编辑被划分为骨架编辑(Skeletal Editing)和外围编辑(Peripheral Editing)。前者修改分子的核心骨架,后者在保持骨架不变的前提下修饰药效团外围。
骨架编辑
而 2025年RSC综述[2]将骨架编辑细分为三类原子级操作:
原子插入(环扩张):代表性方法包括:Levin组使用α-氯二氮杂环丙烷实现吡咯→吡啶和吲哚→喹啉的碳插入[12];Morandi组利用高价碘试剂实现吲哚和吡咯的氮插入[13];Arnold组利用工程化酶实现氮丙啶的对映选择性碳插入[14]。
原子删除(环收缩):Levin组的异头酰胺试剂方法[15]将二级胺转化为异二氮烯中间体,经N₂排出和C–C偶联得到去氮碳环,此工作发表在Nature上,被C&EN专题报道称为"早期里程碑"。此外,光介导的喹啉N-氧化物→吲哚转化[16]和Morandi组的Ni(0)插入内酰胺C–N键[17]也是代表性方法。
原子交换(保持环大小):最具药学价值的是C→N交换("芳香氮扫描"),Levin组[18]通过ipso-选择性氮宾内化策略,在原有碳位置精确安装氮原子。N→C交换方面有Zincke型反应[19]和[4+2]环加成策略[20];Sarpong和McNally & Smith发展了¹⁴N→¹⁵N同位素交换方法。
外围编辑
外围编辑在保持核心骨架的前提下进行位点选择性官能团化。主要策略包括:C–H活化(过渡金属催化和光氧化还原驱动)、原子/官能团交换(氘代丁苯那嗪和氘可来昔替尼已获批上市[1])、官能团迁移(Dong组1,2-羰基迁移法[21],51%收率)、骨架重建和立体化学编辑。
先导化合物优化的四类结构操作
侯廷军课题组2024年JACS综述[22]将结构导向优化任务归纳为四类:
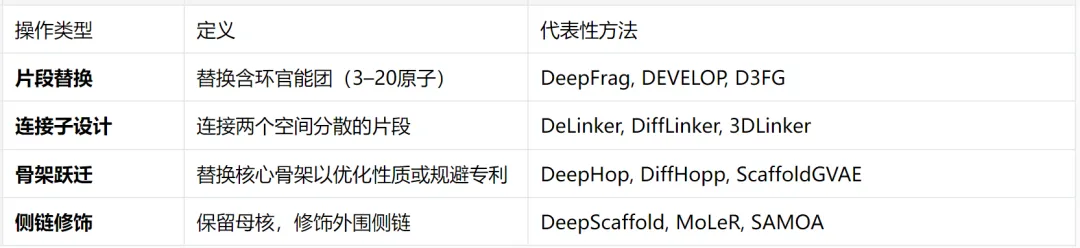
该综述还指出,这些操作的训练数据构建方式天然分化为两类[22]:BRICS和Recap代表规则驱动分解(化学家手工定义切断键类型),MMP分析和自动模板挖掘代表数据驱动分解(从化合物库中统计推导)。以下两章即沿这两个方向展开。
基于规则驱动的分子优化
操作知识来源于专家判断,以显式规则的形式编码。每条操作都有明确的化学依据,可解释性强,但覆盖受限于专家知识的边界。
MolDQN:价键理论的直接编码
MolDQN(Zhou et al., 2019)是最纯粹的规则驱动代表[9],它的操作空间仅由两条化学基本原理定义:元素周期表(哪些原子类型允许)和价键理论(哪些键型是合法的)。
动作空间每步动态生成约10–20个合法动作:原子添加(C/N/O/F/Cl/S,单键或双键)、原子删除(末端原子)、键添加/删除/升级、官能团添加(如–OH)、终止。所有操作执行后经RDKit SanitizeMol验证,无效则奖励-1且状态不变。
由于MolDQN采用隐式氢表示(所有原子添加定义为替换隐式氢),甲基化仅需一个AddAtom(C)动作。但更复杂的变换(如构建环丙烷或骨架编辑)仍需数十步累积。每episode 40步[9]的设置反映了原子层粒度的规划深度需求。
MolDQN使用ε-greedy DQN训练,状态由2048位Morgan指纹(radius=3)编码。其"事后过滤"策略,即 agent采样后才检查价键合法性会导致大量无效采样。
SHARP:BRICS规则驱动的分层片段操作
SHARP[11]的操作空间由BRICS的16条化学键类型规则定义。BRICS覆盖了药物化学中最常见的可逆键连接模式(酯键、酰胺键、醚键等),被切断的键在理论上可通过逆反应重新形成。
两级层次动作。高层动作选择操作类型:片段添加、片段删除、片段替换。低层自回归采样2–3个子动作:对于添加(选择附着原子, 选择片段, 选择片段连接点),对于删除(选择断裂的键),对于替换(选择断裂的键, 选择片段, 选择片段连接点)。
SEM合成可行性掩码。三个预训练掩码模型在RL采样前将不可行操作的概率置零:原子掩码预测原子是否为有效连接点,键掩码预测键是否可断裂,片段掩码预测片段对是否可连接。这种采样前硬约束比MolDQN的事后过滤在效率上有质的提升。
SHARP使用SAC训练,每episode 7步,奖励为Vina分数(0.45)+ 药效团匹配(0.45)+ SASA(0.1)。片段替换在生成中占主导地位。
规则驱动与数据训练的融合。SHARP的操作空间由BRICS规则定义(规则驱动),但SEM掩码的训练数据来自BRICS在ChEMBL34上的分解日志(数据训练)。这是两条路线在实践中融合的典型案例——规则定义"什么操作在概念上可能",数据训练"什么操作在统计意义上可行"。
PGFS:手工筛选和修复的反应模板
PGFS[25]的97条反应模板(15条单分子+82条双分子)来自文献继承和手工修复。与ReACT-Drug的自动推导不同,PGFS的模板是专家逐条筛选的,原模板被手动修复了芳香性和立体化学问题,并按第一反应物角色拆分为方向性模板。
PGFS使用TD3算法训练,f-network选择模板(ECFP4指纹,1024位),π-network以ECFP4为输入、在连续描述符向量空间(RLV2,含QED、MolWt等)输出动作,再经k-NN映射到离散反应物。这种"连续输出→k-NN→离散动作"的设计使确定性策略梯度能用于离散动作选择。
FragDockRL:文献反应规则与商业构建块
FragDockRL[26]的操作空间由两条外部规则定义:58条Hartenfeller等人[37]报道的反应SMIRKS规则,以及Enamine商业构建块目录(124,180个构建块,过滤MW>300后保留113,515个)。
操作定义为"选择一个能与当前分子反应的构建块"。在预处理阶段,每个构建块与58条反应规则预匹配;生成时,系统检索当前分子能参与的反应,仅保留兼容构建块为候选。每episode仅4步——构建块级操作的语义密度极高。
这种设计的思想是:通过与文献反应规则的一致性隐式保证合成可行性,无需额外的SEM训练。但其操作空间完全受限于58条规则对化学反应的覆盖和商业构建块库的规模。
DeepFMPO:基于环原子的片段化规则
DeepFMPO[23]的片段化规则是:断开环原子延伸出的所有单键,形成片段(限制≤12重原子、<4个连接点),从初始分子集构建片段库,片段按相似性组织为平衡二叉树,相似片段在树中相邻。
动作定义为选择片段并翻转其二进制编码中的一个位(仅允许尾部可变位),迫使替换为结构相似的片段,这一设计模拟了研发过程中"微调"先导化合物的方式,DeepFMPO使用双向LSTM的actor-critic架构,奖励基于多目标属性范围的满足程度。
MacFrag:扩展BRICS的规则,打破环键限制
MacFrag[38](Bioinformatics, 2023)代表了规则驱动路线的"规则扩展"方向:不改变规则驱动的根本范式,但通过放松限制条件来扩展覆盖。
传统的BRICS规则不切割环键,这一限制保证了环结构的完整性,但也意味着大环化合物无法被有效片段化。MacFrag的关键创新是去除了BRICS对环键断裂的限制,将可断裂键从16条扩展到49条(通过定义19个原子环境SMARTS组合而成)。同时引入一个可调参数maxSR:所含原子数≤该值的环结构保持不变。当maxSR设为大值时等价于原始BRICS。
MacFrag还引入了一种高效的子图枚举算法(Simple算法)来快速遍历片段空间:将分子转为砌块节点图,枚举指定最大节点数的连通诱导子图,再映射回原始分子提取片段。实验表明,MacFrag比BRICS快2.5–7.9倍,比modified molBLOCKS快11.8–104倍。更重要的是,约13%的MacFrag片段符合RO3(Rule of Three)规则,而modified molBLOCKS和BRICS分别只有5%和6%——环键断裂贡献了82,758个独特的RO3兼容片段。
MacFrag与DigFrag的对比恰好揭示了规则扩展与数据驱动的两条不同路径:MacFrag是通过扩大规则集(16→49条)来提升覆盖,每条规则仍有明确的化学SMARTS定义;DigFrag则是完全放弃规则,用GNN学习"哪里应该切断"。
规则驱动路线的共同优势是可解释性,每条操作都有明确的化学依据。共同局限则是覆盖受专家知识约束,BRICS的16条规则无法覆盖所有化学键切断模式,但MacFrag证明规则扩展(16→49条)可以在保持可解释性的同时显著提升覆盖(RO3片段比例从6%提升到13%)。
基于数据驱动:从数据中统计挖掘操作空间
操作知识来源于对大规模实验数据的统计学习,不需要人来显式定义"什么操作是合法的",而是让数据自动揭示"什么操作在化学实践中发生过"。
ReACT-Drug:mmpdb自动模板挖掘
ReACT-Drug[24]的操作空间不是手工编码的,而是用mmpdb(匹配分子对数据库工具)从ChEMBL中自动提取的。流程为:提取活性分子→mmpdb破碎与索引→识别MMP对→差分转换为反应SMARTS模板→四重过滤(变量≤10原子、变量≤母核50%、核心≥6原子、频率≥1)→保留"数千条"模板。
动作选择使用查询-嵌入匹配架构:ChemBERTa(768维)编码当前分子得到查询向量q_t,对所有候选模板产生的产物分子编码得到动作嵌入e_i,通过Softmax(q_t·e_i)选择。这种架构适用于大规模候选空间——模板数量越多,查询-匹配的优势越明显。ReACT-Drug具有目标不可知性(target-agnostic),策略学习的是一种可跨靶点迁移的"化学转换偏好"。
每episode 15步,奖励为四目标加权:结合亲和力 + 0.1×QED + 0.1×SA + 0.35×新颖性。
与PGFS的对比:两者都使用反应模板,但一个自动挖掘(ReACT-Drug),一个手工筛选(PGFS)。这恰恰体现了数据驱动与规则驱动的核心差异——不在于"是否使用模板",而在于"模板从何而来"。
CReM与FREED:MMP片段互换性的统计学习
CReM[30]是MMP思想的工程实现。其核心操作MUTATE的工作方式是:对分子执行片段分解(默认context_radius=3),将每个片段在预编译ChEMBL/ZINC数据库中查找"在相同化学上下文中出现过的其他片段",返回所有合法的替换分子。
CReM的操作空间完全由数据库统计定义——片段A可以被替换为片段B,不是因为某条规则说"酯键可断裂",而是因为在ChEMBL中确实存在分子对,它们在相同局部环境中分别含有片段A和片段B。GuacaMol基准测试中CReM总分17.919,与Graph GA(17.983)相当。
CReM的一个独特参数是context_radius:radius=0时不考虑任何上下文(类似骨架跳跃),radius=3时保留3键化学环境(类似保守修饰)。这种连续可调的上下文控制是规则驱动框架无法提供的——BRICS的上下文固定为"切断的键类型",而CReM的上下文是数据中观察到的实际化学环境。
FREED[23]使用CReM作为其操作空间定义引擎,分子被CReM分解,保留断键处的位置作为"附着位点"(编码为GNN中的专属节点类型)。FREED的RL架构是单向增长的(只能嫁接片段,不能删除),三层自回归策略选择(连接位点→片段→片段连接点),使用SAC训练。片段库来自ZINC 25万分子的CReM分解(≤12原子,>2频率,66–91片段)。
FREED的核心设计决策单向增长与操作来源无关(CReM本身支持双向MUTATE),它是RL架构层面的选择。这再次说明操作来源和RL设计是正交的。
MMPT-FM与MMPT-RAG:从变换实例到变换规则
MMPT-FM[29]将数据驱动思想推向了最极致的形态:从约263万条MMP实例中提取并去重得到约80万条独特的MMPT(匹配分子对变换),以SMIRKS格式表示。每条MMPT描述了变量A→变量B的映射关系,与分子的常数部分完全解耦。
去重的三重价值:(1)消除上下文噪声,同一变换在不同分子中重复出现不再被多重计数;(2)频率归一化,高频变换(氢→甲基)和罕见变换被赋予相同权重;(3)长尾覆盖,80万条变换覆盖了训练集中仅出现数次的操作。
一个关键发现是MMPT-FM用统一模型处理R基替换和核心跃迁两种药化策略,而REINVENT的专用模块在核心跃迁任务上完全失效(0%召回率)。当操作被定义为上下文无关的变换规则时,骨架级和侧链级别的修改自然统一,它们不过是变量片段大小不同的变换实例。
MMPT-RAG是MMPT-FM的检索增强扩展。核心创新在于允许用户提供一个项目特异性的参考分子集,系统检索与当前输入变量v_A结构相似的已知MMPT,将生成分布从"全局化学先验"引导至"项目特异性偏好"。理论证明MMPT-RAG的分布是全局先验和参考分布在凸组合之间的插值:
检索到的输出变量经HDBSCAN聚类后,以最大公共子结构(MCS)作为掩码模板,指导MMPT-FM在特定子空间内生成。这使得同一框架既可以用于广谱探索(关闭检索),也可以用于聚焦优化(开启检索)。
Modof:分子对差异编码的数据驱动修改
Modof代表了数据驱动路线的另一种形态:不预定义任何规则来指定"在哪里修改"或"如何修改",而是从超过10万条分子对(优化前Mx→优化后My)的训练数据中学习差异模式。
Modof的核心是差异编码器:通过图消息传递网络和树消息传递网络分别处理分子图和连接树,将"Mx和My之间的差异"编码为潜向量z。修改过程是序列化的:(1)断开位点预测,为连接树上每个节点打分,最高分节点为断开位点;(2)移除片段预测,判断断开位点连接的哪个片段应被移除;(3)新片段附着,迭代决定新片段的类型和连接点。整个过程由化学价键检查保证合法性。
Modof的操作知识完全来自数据,断开位点的选择、移除片段的判断、新片段的生成,都不是任何手工规则的产物,而是从分子对数据中统计学习的结果。与CReM(显式存储片段互换性数据库)不同,Modof将操作知识隐式编码在神经网络的参数中。
DigFrag:GNN注意力驱动的数字化片段分割
DigFrag[39](Nature Communications Chemistry)代表了数据驱动路线的一种新形态:用GNN的图注意力机制自动学习"在哪里切断分子",完全不需要预定义断键规则。
DigFrag的工作流程是:将分子图G=(V,E)输入图注意力网络,通过多层注意力层为每个原子学习嵌入表示,聚合为"超级节点",再通过进一步注意力处理得到片段的嵌入表示。模型的训练目标是精准分割药物和农药片段——训练数据来自PADFrag数据库(收录FDA批准的1,652种药物和1,259种商业农药)。
与传统方法的对比揭示了DigFrag的关键特征:DigFrag分割的片段与BRICS、RECAP和MacFrag的重叠率仅为9.97%–21.37%(药物)和8.94%–15.20%(农药),表明它能发现传统规则无法捕获的独特片段。在结构多样性方面,DigFrag片段的可旋转键数量更多,这可能归因于其独特的环状结构断裂方式。t-SNE可视化显示,DigFrag在0.4和0.6相似性阈值下的聚类比率最高,表明其片段具有更高的结构多样性。
基于DigFrag片段训练的DeepFMPO模型在MOSES基准上Filters得分达到0.828,优于基于BRICS、RECAP和MacFrag片段的模型。这揭示了一个有趣的现象:AI模型更偏好AI来源的训练数据——数据驱动的片段化可能产生更适合深度生成模型的片段分布。经过筛选,研究最终确定了24个药物分子和20个农药分子(QED>0.75、SA<3)。
DigFrag与CReM和Modof形成了数据驱动路线内部的三个子类别:CReM通过统计片段共现模式定义操作,Modof通过学习分子对的差异编码定义操作,DigFrag通过学习"哪里是片段的自然边界"定义操作。三者都不依赖化学家手工编码的断键规则,但对"数据中的什么信号定义了操作"给出了不同的回答。
数据驱动路线的共同优势是覆盖广:80万条变换远超手工枚举数量,自动模板挖掘捕获手工遗漏的反应类型,DigFrag甚至发现了与BRICS重叠率仅10%的独特片段空间。共同局限是质量依赖数据:数据偏倚会被继承,未出现的变换无法生成。但DigFrag揭示了一种自我强化的可能性——AI生成的片段反而更适合AI模型的训练。
强化学习:跨越两条路线的统一优化引擎
无论操作空间由规则还是数据定义,RL作为优化引擎的设计选择是通用的。
MDP建模
所有使用强化学习的分子优化框架都建模为马尔可夫决策过程(MDP),但在终止条件上存在差异:
固定episode长度:SHARP(7步)、ReACT-Drug(15步)、FragDockRL(4步),可能过早截断或浪费步数。可变长度+Stop动作:MolDQN(≤40步+Stop)、FREED(生长至Stop或达到终止条件),比较灵活,但需要学习何时终止。固定变换步数:DeepFMPO(固定步数内逐步微调),约束了探索范围但简化了训练。
操作来源对此维度无直接影响,同一条CReM片段库既可以嵌入固定episode(如SHARP风格)也可以嵌入可变长度(如FREED风格)的MDP。
约束策略
化学合理性的保证有四种约束时机:

SHARP的经验表明:采样前硬约束的效果远优于奖励项软约束——RL的信用分配难以精确地将合成不可行性归因到具体操作上。
操作来源与约束策略之间存在一个微妙的关系:规则驱动框架更容易实现硬约束(BRICS规则天然定义了"什么操作可能"),而数据驱动框架的约束更多依赖统计阈值(如CReM的min_freq参数)。但在实践中,两者都可以通过组合策略实现多层防护。
多智能体与多阶段RL:CSstep的扩展
CSstep[40](Chemical Engineering Science, 2025)在MolDQN的原子级动作空间基础上,对RL架构做了两个创新扩展:
多智能体联合决策。不再用单一奖励项整合多个目标(如活性、SA、QED的加权和),而是为每个优化目标分配独立的DQN agent。在每一步,所有agent各自输出Q值,动作选择基于Q值的总和。这种设计使多目标之间的权衡由agent的联合决策自然涌现,而非依赖人工调参的权重方案。
多阶段短链训练。不再使用一条长MDP链(容易导致agent"迷失方向"),而是将优化过程拆分为多个短阶段。每个阶段运行短episode后,最佳分子成为下一阶段的新起点,但agent的神经网络参数被保留并持续更新,知识随阶段累积而不遗忘。
CSstep的贡献不在操作空间的定义(它沿用MolDQN的价键规则),而在RL架构对多目标和长链问题的适应能力。这提醒我们,操作来源的选择只是分子优化系统设计的一个维度,RL架构的创新同样重要。
非RL优化方法:遗传算法、元启发式与进化搜索
虽然本文聚焦于RL,但值得简要对比遗传算法(GA)和元启发式方法,它们同样可以搭载规则驱动或数据驱动的操作空间。
GA方法(如MolFinder、STONED、GB-GA-P)通过群体搜索和遗传算子(交叉、变异)来探索化学空间。其"变异算子"本质上也是分子编辑操作。与RL的关键区别在于:GA不学习"何时应用何种操作"的策略,而是依赖随机变异和自然选择。
STELLA[42](Scientific Reports, 2025)是元启发式方法的最新代表。其FRAGRANCE模块从约2,300万ChEMBL/SureChEMBL化合物中构建了约260万独特片段的数据库(采用环-连接子分解+BRICS束搜索)。三种变异操作——FRAGRANCE突变(k-NN片段替换)、MCS交叉和修剪——均在片段级别而非原子级别执行。STELLA采用聚类引导的构象空间退火(CSA),通过逐步收紧的聚类距离阈值实现从全局探索到局部利用的平滑过渡。在Abl1和p53靶点上,STELLA分别生成251和210个分子簇,而REINVENT4仅生成42和61个。
LLM驱动的进化搜索:ICLR 2025的LLM-EVO框架[41]将LLM作为变异算子生成器,根据当前分子和优化目标动态提出结构修改建议。这一方向模糊了"操作来源"的传统边界——LLM的化学知识既来自训练数据(数据驱动),又在推理时受prompt中的化学规则约束(规则驱动)。
MolAct:当引入 LLM
MolAct[43]引入了一种超越"规则驱动 vs 数据驱动"二分的新范式:LLM作为策略,动作由LLM在推理时动态组合,工具作为化学合理性验证器。
MolAct将分子编辑建模为工具增强的多步决策过程。在每一步,LLM策略可以:(1) 推理(内部"思考"),(2) 调用外部工具(化学合理性验证、相似度检查、性质预测),(3) 应用编辑操作(在工具验证的位点上进行官能团的添加/删除/替换),(4) 终止。
训练采用两阶段课程。阶段一(MolEditAgent):仅训练编辑技能,学习如何有效使用工具和编辑操作而不涉及性质优化目标。阶段二(MolOptAgent):在阶段一的基础上引入性质优化奖励(LogP、溶解度、DRD2/JNK3/GSK3β活性等)。实验表明,直接从优化开始训练(单阶段)的成功率接近零,因为模型从未学会有效的工具使用和终止策略。
MolAct使用GRPO(Group Relative Policy Optimization)训练,对每个prompt生成K条并行轨迹,优势在组内相对计算。梯度仅作用于agent自身产生的token,工具输出和prompt作为固定上下文。
MolAct的操作空间既不是手工规则预定义的(非规则驱动),也不是从数据库中统计挖掘的(非数据驱动),而是由LLM在工具反馈的实时约束下动态构成的。这在我们的分类体系中打开了一个新维度:操作知识的来源可以是"基础模型参数中的隐式化学知识 + 工具的显式化学约束"。
仍在路上
两条路线的融合趋势
最前沿的工作正趋向于融合规则与数据。SHARP的SEM掩码(在规则生成的片段上训练)是最早的范例。MMPT-RAG更进一步:全局变换库(数据驱动)通过项目参考集(可能包含化学家手工标记的优先变换)进行检索增强,使生成分布从"化学上合理"转向"项目上推荐"。
一个值得关注的方向是在线闭环融合:RL agent在执行过程中发现当前操作库无法完成的变换,触发数据挖掘模块从ChEMBL中搜索对应的MMP实例来扩展操作空间,这一流程尚未在任何已发表框架中实现。
合成路线gap的桥接:从事后过滤到直接优化
RL生成的分子编辑操作序列与实际合成路线之间的gap是本文反复提及的开放问题。2025年出现了两个具体解决方案。
SynTwins[44](RSC Chemical Science, 2025)采取"先逆向合成分析、再搜索类似构建块"的策略。给定目标分子,先通过多步逆合成模板(深度≤3)将其分解为前体构建块,然后用k-NN(ECFP指纹)搜索结构相似且商业可得的替换构建块,最后通过正向反应模板组合出k²个分子类似物。这一流程完全不需要ML训练。在嵌入REINVENT的优化循环时(Syn-Model模式),每一步生成都经逆合成验证——虽然峰值分数略降(Δ≈0.35-0.50),但每个分子都附带完整合成路线。在100个FDA批准药物上精确匹配率达17%(基线方法仅1%)。
Guo & Schwaller[45](RSC Chemical Science, 2025)采取相反的思路:直接将逆合成模型作为RL奖励函数的一部分。使用Mamba架构的自回归语言模型Saturn作为生成器,采样效率足以在仅1,000次oracle调用内完成优化(传统方法需32,000–256,000次)。关键发现:SA分数作为合成可行性的代理指标,在药物类分子空间中与逆合成可解性高度相关,但在功能材料等非药物空间中相关性急剧下降——此时直接优化逆合成模型显著更优。
这两个工作共同指向一个趋势:合成可行性的保证正在从"预定义规则过滤"(BRICS、反应模板)向"在线逆合成验证"演进。这与操作来源的演进是平行的——规则驱动的操作空间天然倾向于可合成操作但覆盖有限;数据驱动的操作空间覆盖广但可能生成不可合成分子;在线逆合成验证提供了第三条路:既保留宽覆盖,又实时保证合成可行性。
LLM动态组合的操作空间
MolAct[43]和ChemCRAFT[33]代表了超越"规则驱动 vs 数据驱动"二元分类的新范式。在这些框架中,操作空间不再预先定义,既不是化学家手工编码的,也不是从数据库中统计挖掘的,而是由LLM在推理时动态构成的。LLM的参数编码了来自预训练数据的隐式化学知识,而外部工具(化学验证器、性质预测器)提供实时的显式约束。这打破了本文"操作来源决定一切"的前提假设,将问题从"谁定义了操作空间"转向"谁在实时构建操作空间"。ChemCRAFT[33]将动作定义为化学工具API调用,LLM作为推理引擎。这一范式下,操作知识被编码在LLM参数中(数据驱动),但LLM也可以遵循化学家提供的约束规则(规则驱动),代表了两种路线在更高抽象层次上的融合。
MMP变换的方向性问题
MMP天然是有向的,一条记录只证明"在某个分子对中,片段A变成了片段B",不保证反向变换也存在。这对双向RL的MDP遍历性构成直接挑战:如果agent通过v_A→v_B从状态S1走到S2,但v_B→v_A在变换库中不存在,agent就无法原路返回。这个问题在实践中有四条解决路径,各有不同的适用场景:
上下文互换(CReM方案):CReM不存储有向变换,而是统计"在相同化学上下文中出现过哪些片段"。任何两个在相同上下文中出现过的片段都可以互换,天然双向。代价是丢失了变换的方向信息(无法区分A→B比B→A更常见)。对称增强:为每条v_A→v_B人工生成反向v_B→v_A。80万条变成160万条。问题在于化学上并非所有变换都可以逆转(如氧化反应),人工反向会引入虚假操作。混合操作空间:正向探索用MMPT(覆盖面大),反向回退用规则驱动操作(BRICS切断、CReM DELETE)。FREED选择单向增长的深层原因很可能就在于此,不是CReM不支持双向,而是设计者倾向于把反向操作交给更保守的规则引擎。差异编码(Modof方案):不显式存储A→B变换,而是学习分子对差异的向量表示。差异编码在数学上可近似对称,"A和B的差异"≈负的"B和A的差异",这比显式存储有向变换更接近可逆。需要注意的是,这个问题在实际中的影响比理论上轻。大多数药物化学中的MMP变换在统计上近似可逆(甲基→乙基和乙基→甲基通常都有记录),真正的不可逆变换比例不高。而且目前主流的片段级框架(FREED、STELLA等)实际用的是CReM而非纯MMPT,CReM的上下文共现机制天然规避了方向性问题。真正的隐患集中在MMPT-FM这类大规模有向变换库直接接入双向RL的场景。
参考文献
Rational Molecular Editing: A New Paradigm in Drug Discovery. J. Med. Chem.2024, 67 (14), 11459–11466. DOI: 10.1021/acs.jmedchem.4c01347 Remodelling molecular frameworks via atom-level surgery: recent advances in skeletal editing of (hetero)cycles. Sharma R, Arisawa M, Takizawa S, Salem MSH. Org. Chem. Front.2025, 12 (5), 1633–1670. DOI: 10.1039/d4qo02157f Revolutionizing Playing with Skeleton Atoms: Molecular Editing Surgery in Medicinal Chemistry. ACS Pharmacol. Transl. Sci.2025, 8 (2), 190–195. PMCID: PMC11851142 Single-atom logic for heterocycle editing. Jurczyk J, Woo J, Kim SF, Dherange BD, Sarpong R, Levin MD. Nature Synthesis2022, 1, 352–364. DOI: 10.1038/s44160-022-00052-1 A review of reinforcement learning in chemistry. Gow S, Niranjan M, Kanza S, Frey JG. Digital Discovery2022, 1, 543–554. DOI: 10.1039/d2dd00047d Optimizing Drug Discovery: How Reinforcement Learning Transforms Molecular Design and Property Prediction. MolEngSci2024 (blog post, no DOI). Molecular Design in Synthetically Accessible Chemical Space via Deep Reinforcement Learning. ACS Omega2021, 6 (1), 424–433. DOI: 10.1021/acsomega.0c04153 Sample efficient reinforcement learning with active learning for molecular design. Chemical Science2024, 15 (11), 4146–4160. DOI: 10.1039/d3sc04653b Optimization of Molecules via Deep Reinforcement Learning. Zhou Z, Kearnes S, Li L, Zare RN, Riley P. Scientific Reports2019, 9, 10752. DOI: 10.1038/s41598-019-47148-x REINVENT4: Modern AI-driven generative molecule design. ChemRxiv2023. DOI: 10.26434/chemrxiv-2023-xt65x. GitHub: github.com/MolecularAI/REINVENT4 SHARP: Generating Synthesizable Molecules via Fragment-Based Hierarchical Action-Space Reinforcement Learning for Pareto Optimization. Kim J, Ryu S, et al. J. Chem. Inf. Model.2025. DOI: 10.1021/acs.jcim.5c01699 Carbon Atom Insertion into Pyrroles and Indoles Promoted by Chlorodiazirines. Dherange BD, Kelly PQ, Liles JP, Sigman MS, Levin MD. J. Am. Chem. Soc.2021, 143 (30), 11337–11344. DOI: 10.1021/jacs.1c06287 Late-stage diversification of indole skeletons through nitrogen atom insertion. Reisenbauer JC, Green O, Franchino A, Finkelstein P, Morandi B. Science2022, 377 (6610), 1104–1109. DOI: 10.1126/science.add1383 Biocatalytic One-Carbon Ring Expansion of Aziridines to Azetidines via a Highly Enantioselective [1,2]-Stevens Rearrangement. Miller DC, Lal RG, Marchetti LA, Arnold FH. J. Am. Chem. Soc.2022, 144 (11), 4739–4745. DOI: 10.1021/jacs.2c00251 Skeletal editing through direct nitrogen deletion of secondary amines. Kennedy SH, Dherange BD, Berger KJ, Levin MD. Nature2021, 593 (7858), 223–227. DOI: 10.1038/s41586-021-03448-9 Scaffold hopping by net photochemical carbon deletion of azaarenes. Woo J, Christian AH, Burgess SA, Jiang Y, Mansoor UF, Levin MD. Science2022, 376 (6592), 527–532. DOI: 10.1126/science.abo4282 Skeletal metalation of lactams through a carbonyl-to-nickel-exchange logic. Zhong H, Egger DT, Morandi B. Nat. Commun.2023, 14, 5523. DOI: 10.1038/s41467-023-40979-3 Aromatic nitrogen scanning by ipso-selective nitrene internalization. Pearson TJ, Shimazumi R, Dherange BD, et al. Science2023, 381 (6665), 1474–1479. DOI: 10.1126/science.adj5331 Zincke-type N→C transmutation (secondary citation). Original works: Kano N et al. (2021, Tetrahedron Lett.); Morofuji T et al. (2021, Chem. Sci.); Glorius F et al. (2024, J. Am. Chem. Soc.). (引自综述[2]) [4+2] cycloaddition strategy for pyridine→benzene (secondary citation). Original work: Studer A et al. (2024, J. Am. Chem. Soc.). (引自综述[2]) 1,2-carbonyl migration for steroid modification (secondary citation). Original work: Dong G et al. (2021, Nat. Chem.). (引自综述[1]) Deep Lead Optimization: Leveraging Generative AI for Structural Modification. Hou T, et al. J. Am. Chem. Soc.2024, 146 (46), 31357–31370. DOI: 10.1021/jacs.4c11686 Hit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation (FREED). Yang S, Hwang D, Lee S, Ryu S, Hwang SJ. In NeurIPS 2021, 7924–7936. Also arXiv:2110.01219. DOI: 10.48550/arXiv.2110.01219 ReACT-Drug: Reaction-Template Guided Reinforcement Learning for de novo Drug Design. arXiv2025, arXiv:2512.20958. DOI: 10.48550/arXiv.2512.20958 Learning To Navigate the Synthetically Accessible Chemical Space Using Reinforcement Learning. In ICML 2020. Also arXiv:2004.12485. DOI: 10.48550/arXiv.2004.12485 FragDockRL: A Reinforcement Learning Framework for Fragment-Based Ligand Design via Building Block Assembly and Tethered Docking. bioRxiv2025. DOI: 10.1101/2025.08.12.670002 Matched Molecular Pair Analysis in Short: Algorithms, Applications and Limitations. Comput. Struct. Biotechnol. J.2017, 15, 86–90. DOI: 10.1016/j.csbj.2016.12.003 Matched Molecular Pair Analysis in Drug Discovery: Methods and Recent Applications. Shi S, Fu L, Hou T, Cao D, et al. J. Med. Chem.2023, 66 (13), 8287–8301. DOI: 10.1021/acs.jmedchem.2c01787 MMPT-RAG: Retrieval-Augmented Foundation Models for Matched Molecular Pair Transformations to Recapitulate Medicinal Chemistry Intuition. arXiv2026, arXiv:2602.16684. DOI: 10.48550/arXiv.2602.16684 CReM: Chemically Reasonable Mutations (version 0.2.17). GitHub: github.com/DrrDom/crem; PyPI: crem 0.2.17. DiffHopp: A Graph Diffusion Model for Novel Drug Design via Scaffold Hopping. Torge J, Harris C, Mathis SV, Lió P. arXiv2023, arXiv:2308.07416. DOI: 10.48550/arXiv.2308.07416 3DLinker: An E(3) Equivariant Variational Autoencoder for Molecular Linker Design. Huang Y, Peng X, Ma J, Zhang M. In ICML 2022. Also arXiv:2205.07309. ChemCRAFT: Agentic Reinforcement Learning for Molecular Design. arXiv2026, arXiv:2601.17687. DOI: 10.48550/arXiv.2601.17687 MMPT-RAG: Retrieval-Augmented Foundation Models for Matched Molecular Pair Transformations to Recapitulate Medicinal Chemistry Intuition. arXiv2026, arXiv:2602.16684. DOI: 10.48550/arXiv.2602.16684 Modof: A deep generative model for molecule optimization via one fragment modification. Chen Z, Min MR, Parthasarathy S, Ning X. Nature Machine Intelligence2021, 3, 1047–1056. DOI: 10.1038/s42256-021-00410-2 Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design (DeepFMPO). Ståhl N, Falkman G, Karlsson A, Mathiason G, Boström J. J. Chem. Inf. Model.2019, 59 (7), 3166–3179. DOI: 10.1021/acs.jcim.9b00325 A Collection of Robust Organic Synthesis Reactions for In Silico Molecule Design. Hartenfeller M, Zettl H, Walter M, et al. J. Chem. Inf. Model.2011, 51 (12), 3093–3098. DOI: 10.1021/ci200379p MacFrag: segmenting large-scale molecules to obtain diverse fragments with high qualities. Diao Y, Hu F, Shen Z, Li H. Bioinformatics2023, 39 (1), btad012. DOI: 10.1093/bioinformatics/btad012 DigFrag as a digital fragmentation method used for artificial intelligence-based drug design. Yang R, Zhou H, Wang F, Yang G. Commun. Chem.2024, 7, 258. DOI: 10.1038/s42004-024-01346-5 CSstep: Step-by-step exploration of the chemical space of drug molecules via multi-agent and multi-stage reinforcement learning. Chem. Eng. Sci.2025, 317, 122048. DOI: 10.1016/j.ces.2025.122048 Efficient Evolutionary Search over Chemical Space with Large Language Models. Wang H, et al. In ICLR 2025. Also arXiv:2406.16976. STELLA: A drug design framework enabling extensive fragment-level chemical space exploration and balanced multi-parameter optimization. Sci. Rep.2025, 15, 28135. DOI: 10.1038/s41598-025-12685-1 MolAct: An Agentic RL Framework for Molecular Editing and Property Optimization. arXiv2025, arXiv:2512.20135. DOI: 10.48550/arXiv.2512.20135 SynTwins: A retrosynthesis-guided framework for synthesizable molecular analog generation. Chem. Sci.2025. DOI: 10.1039/d5sc05225d Directly optimizing for synthesizability in generative molecular design using retrosynthesis models. Guo J, Schwaller P. Chem. Sci.2025. DOI: 10.1039/d5sc01476j
