 夜雨聆风
夜雨聆风
文献来源: Hu W, Sun Y, Li T, et al. The evolution of computation-driven paradigms in targeted peptide drug design: From predictive modeling to generative AI and clinical translation. The Innovation Drug Discovery 1(1): 100009, 2026.DOI: 10.59717/j.xinn-drugdisc.2026.100009
一、引言:多肽药物的独特治疗定位
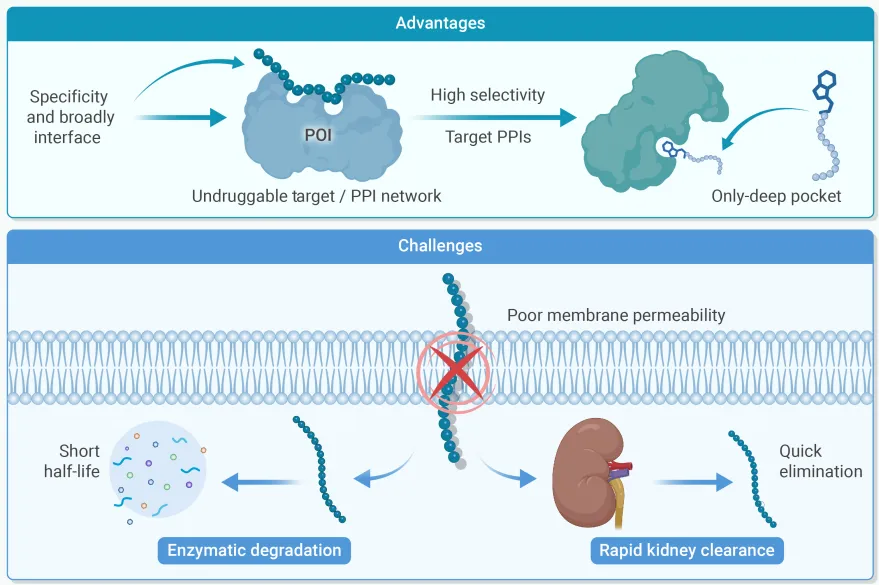
多肽在化学本质上定义为2至50个氨基酸残基构成的链状分子,在治疗学上占据着小分子药物与大分子生物制剂之间的独特生态位。与小分子药物相比,多肽具有更大的结合界面面积(400–1000 Ų),可与传统小分子无法结合的平坦生物界面发生高选择性相互作用,从而靶向此前被认为"不可成药"的靶点。
尤为突出的是,多肽在靶向胞内蛋白-蛋白相互作用(PPI)网络方面展现出显著优势。相较于单克隆抗体,多肽还具有组织穿透力更强、免疫原性潜在更低、制造成本更低等特点。GLP-1受体激动剂(利拉鲁肽、司美格鲁肽)在代谢与心血管疾病治疗中的革命性成功,已将多肽确立为现代制药工业的核心支柱之一。
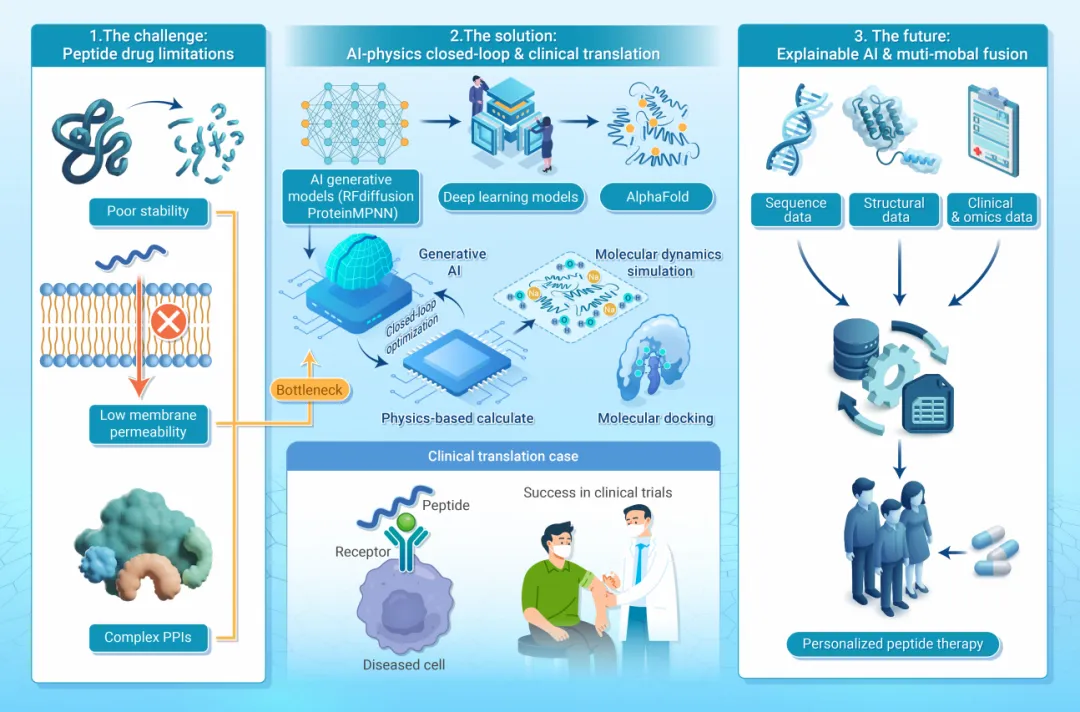
二、多肽药物的药代动力学瓶颈
尽管治疗潜力巨大,多肽的临床转化长期受制于以下系统性药代动力学障碍:
2.1 快速酶解降解
多肽进入血液循环后,迅速被DPP-IV等蛋白酶识别并降解,导致活性半衰期极短(有时仅数分钟),难以维持有效治疗浓度。
2.2 肾脏快速清除
多肽分子量相对较小,可被肾小球高效滤过,导致血浆功能半衰期极短,成为维持稳态血药浓度的主要障碍。
2.3 膜渗透性差
多肽固有的亲水性和较大分子尺寸严重限制其穿越生物膜的能力,口服制剂的生物利用度问题尤为突出。
2.4 序列空间优化困境
传统药物化学依赖骨架环化(构象约束)、非天然氨基酸引入(抗酶解)、PEG化(延长循环)等经验性修饰策略。这些试错方法耗时耗力,且难以在活性、稳定性与膜渗透性之间高效寻优。
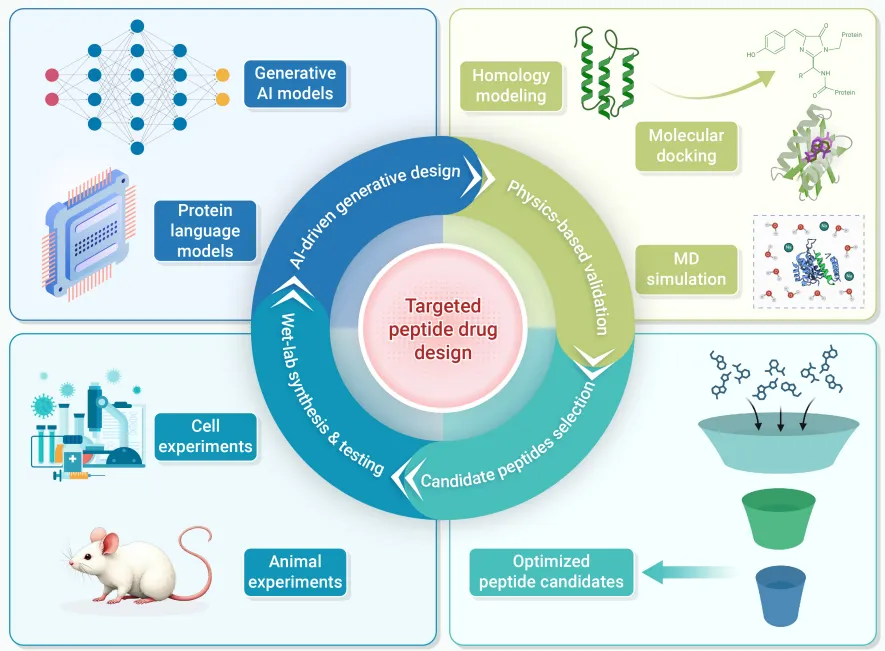
三、主流分子建模方法
多肽药物发现的理性设计依赖三类基础计算工具协同运作。
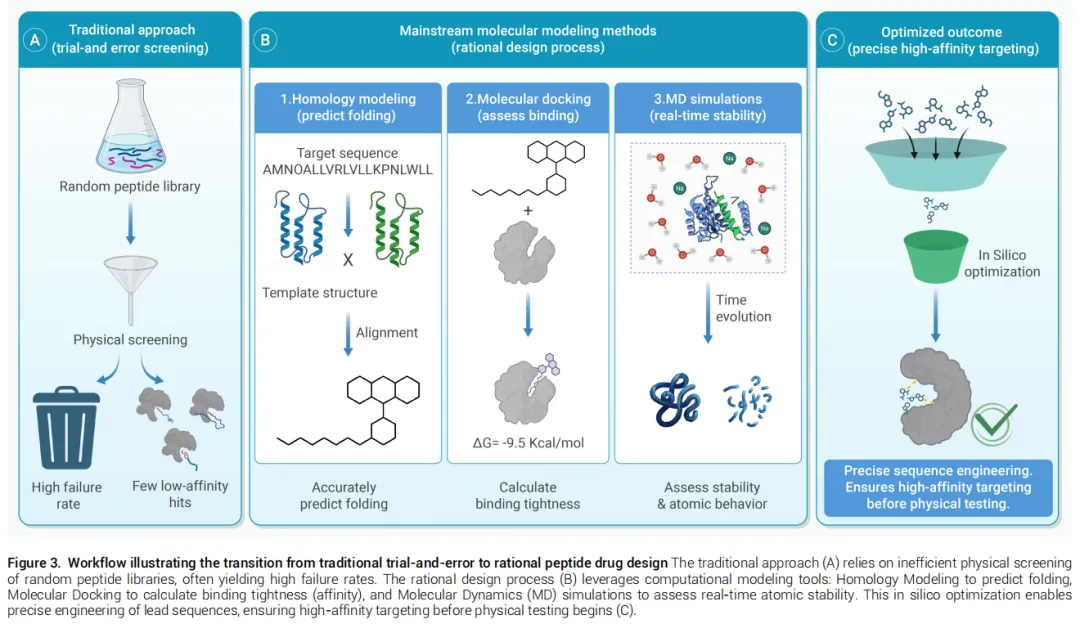
3.1 同源建模
同源建模基于"序列相似的蛋白倾向于折叠成相似三维结构"这一进化规律,利用已知模板预测未知靶标的三维结构。
核心工作流程: 模板选择 → 序列比对 → 模型精修
关键精度阈值: 当目标序列与模板序列一致性超过30%时,主链构象和结构基序的可靠性显著提升。
在PPI靶点中的特殊价值: 基于竞争性多肽设计逻辑,同源建模可重建靶蛋白与天然结合伴侣的复合物结构,提取界面热点残基(对结合自由能贡献不成比例的残基),进而精确映射平坦生物界面,指导MDM2/MDMX、BCL-2家族等靶点抑制剂的设计。
局限性: 高度柔性环区和无序结构域的建模精度较低;序列差异与结构偏差之间的非线性关系要求严格的多模型比较和验证。
3.2 分子对接
分子对接通过模拟多肽-蛋白结合构象并量化相互作用强度,为先导化合物筛选和优化提供结构依据。
多肽对接的核心挑战:多肽骨架和侧链均具有高度内禀柔性,构象空间远大于小分子,对采样算法的搜索能力构成严峻挑战。
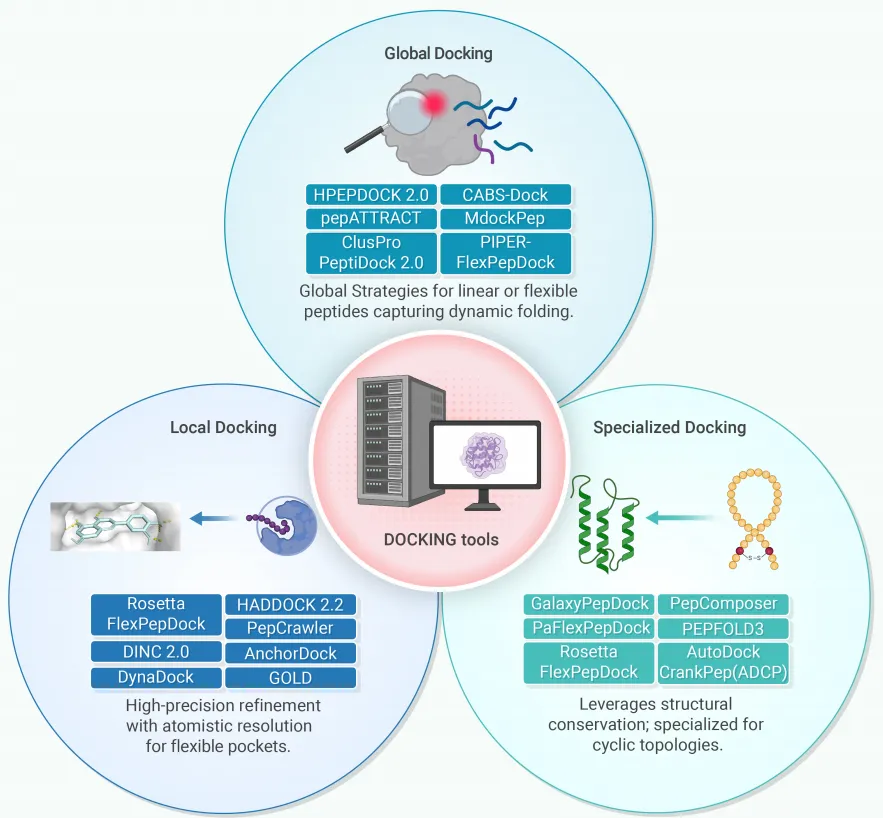

对接策略分类(覆盖精度-效率权衡):
分层对接策略(Ciemny等推荐): 先用粗粒化全局算法捕获结合热点,再用高精度局部工具优化界面原子接触,有效兼顾采样效率与结构精度。
新兴范式 — AI原生扩散框架(RAPiDock): 通过渐进式去噪随机坐标导航高维能量景观,克服传统方法陷入局部极小的缺陷,作为物理精修协议的有力补充。
转化案例: 口服PCSK9抑制剂MK-0616的发现中,计算分析指导了大环支架的刚性化设计,使高效力与肠道渗透性得以共存。
3.3 分子动力学模拟
分子动力学(MD)模拟在时间维度上补充对接预测,通过求解经典力场下的原子运动方程,捕捉溶剂环境中多肽-蛋白体系的动态演化,直接观测折叠路径、构象转变和相互作用持久性。
关键参数选择:
• 显式溶剂模型: 氢键网络和水化效应表征精准,但计算代价高 • 隐式溶剂模型: 计算效率高,但精细相互作用的精度受限
增强采样技术(克服能量景观粗糙性):
• REMD(副本交换分子动力学): 多温度副本并行运行,借助温度梯度跨越能量垒,已在淀粉样肽结构表征中证明有效 • aMD + 元动力学混合策略: aMD负责快速构象扫描,元动力学负责精确自由能估算,联合使用提升预测精度 • ML引导MD: Weber等证明机器学习算法可高效引导MD探索关键构象区域,加速高维构象采样的收敛
典型应用: Lau等利用MD模拟验证司美格鲁肽亲水连接臂的柔性足以容纳白蛋白结合,同时不干扰GLP-1部分与受体的关键相互作用,从而确保药代工程不损害治疗效力。
四、机器学习与AI驱动的多肽设计
4.1 预测式AI:从序列到结构与功能
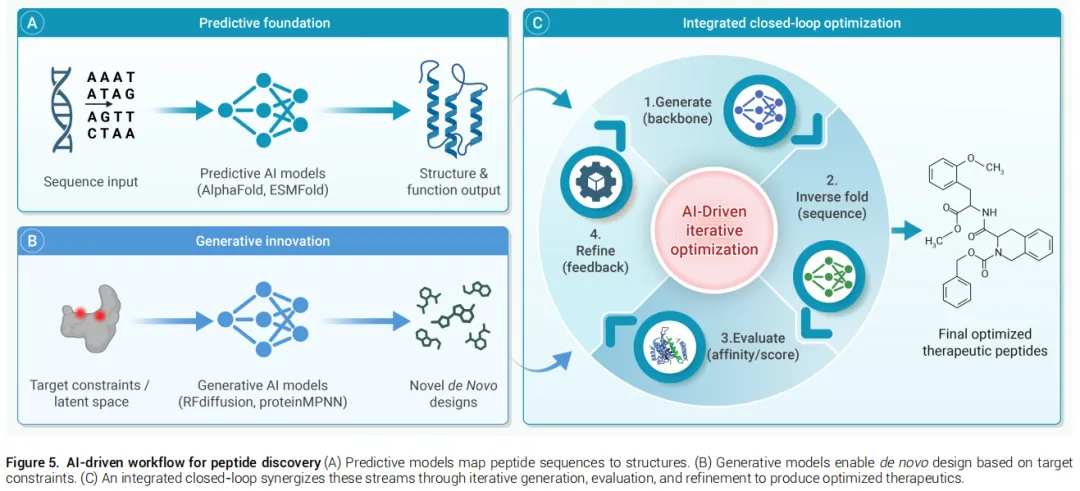
AlphaFold系列
AlphaFold利用多序列比对(MSA)提取进化信息,结合注意力机制捕获残基间长程关联,实现近实验精度的结构预测。其在多肽-蛋白复合物建模和结合模式评估中尤为强大,有效应对传统对接算法难以处理的诱导契合构象变化。
关键局限: 对MSA数据的依赖构成内禀瓶颈——针对de novo设计的人工序列或缺乏同源数据的孤儿多肽,预测置信度显著下降。
AlphaFold 3的突破: 通过广义全原子表示替代严格的残基级别标记化,原生支持修饰残基和非标准配体,是建模非天然氨基酸的重大进展。
ESMFold(蛋白质语言模型路线)
ESMFold基于Meta AI的ESM-2蛋白质语言模型,直接从单条序列预测结构,无需计算密集的多序列比对,速度比AlphaFold快约一个数量级,非常适合需要快速评估数百万候选多肽的高通量筛选。
分层预测策略(业界共识):
• 第一层(大规模初筛): ESMFold — 毫秒级推理,快速过滤百万量级序列库 • 第二层(先导优化): AlphaFold — 高保真原子坐标,精确表征界面位阻
4.2 生成式AI:从头设计与化学空间探索
本文综述提出基于主要数据模态的三类生成框架分类:
(1)序列生成优先范式
代表模型:变分自编码器(VAE)、生成对抗网络(GAN)、ProGen2 / ProtGPT2、EvoDiff
工作机制:将离散氨基酸序列映射至连续潜空间,从流形采样生成覆盖抗菌肽、细胞穿透肽等功能多肽的多样候选序列,超越天然数据库的多样性边界。
优势: 对于三维结构动态或不严格定义的功能多肽尤为有效。
代表工具:
• EvoDiff(微软): 在序列空间直接扩散生成,无需三维结构模板,对本征无序区域和柔性线性多肽尤具优势 • PepMLM: 基于掩码语言模型,完全绕过三维结构依赖,直接生成靶向结合多肽序列
(2)结构生成优先范式
代表模型:RFdiffusion、RFdiffusion3、BindCraft
工作机制:以靶蛋白结合口袋为条件输入,通过渐进式去噪随机坐标从头生成与靶标界面精确互补的多肽骨架,优先保证几何互补性。
决定性意义: 针对平坦PPI界面等"不可成药"位点,精确结构匹配是结合的先决条件,此范式提供了最直接的解决路径。
BindCraft(端到端自动化流程): 整合RFdiffusion和ProteinMPNN,内置置信度过滤,显著降低线性及结构约束结合体设计的门槛。
(3)序列-结构协同优化范式
代表模型:ODesign
将多模态生物分子相互作用统一在单一生成框架内,而非将靶标视为静态约束,捕获多肽与靶标之间的相互结构适应,向多模态、相互作用感知的生成模型方向快速演进。
4.3 逆折叠:从三维骨架到可合成序列
生成扩散模型输出的三维骨架必须经过**逆折叠(inverse folding)**步骤转化为实际可合成的氨基酸序列。
| ProteinMPNN | ||
| ESM-IF1 | ||
| LigandMPNN |
"结构幻觉"问题: 当前扩散模型易生成几何上与靶点互补但违反基本理化原则的骨架——孤立的二级结构、缺失的疏水核心或过度溶剂暴露的疏水表面。这类幻觉骨架在体外验证中因聚集、溶解度差或无法稳定折叠而失败。ProteinMPNN等工具的核心价值正在于此:作为连接几何设计与化学现实的必要过滤器。
4.4 ADMET与成药性评估
体外高亲和力≠体内治疗效果。 多肽候选药物临床失败的主要驱动因素是ADMET性质缺陷。
关键预测工具:
整合策略: 单纯依赖序列预测器对高度新颖的线性支架可能产生假阳性。前沿工作流将一维序列预测器与三维结构和动态验证深度整合——候选序列通过初筛后,先经AlphaFold预测结构,再经GROMACS进行显式水环境中的MD验证,动态区分物理稳定序列与聚集倾向序列。
五、闭环设计框架:整合AI与物理验证
综述提出的闭环工作流将生成式与预测式AI统一在迭代优化框架中:
生成高质量骨架(RFdiffusion) ↓逆折叠恢复序列(ProteinMPNN/LigandMPNN) ↓结构置信度评估(AlphaFold:PAE / ipTM指标) ↓多目标属性预测(ADMET / 溶解度 / 膜渗透性) ↓硬规则过滤(分子量、聚集倾向等阈值) ↓物理验证层级(能量最小化 → 短时MD → 自由能计算) ↓反馈信号更新生成模型分布参数 ↓(循环迭代直至收敛)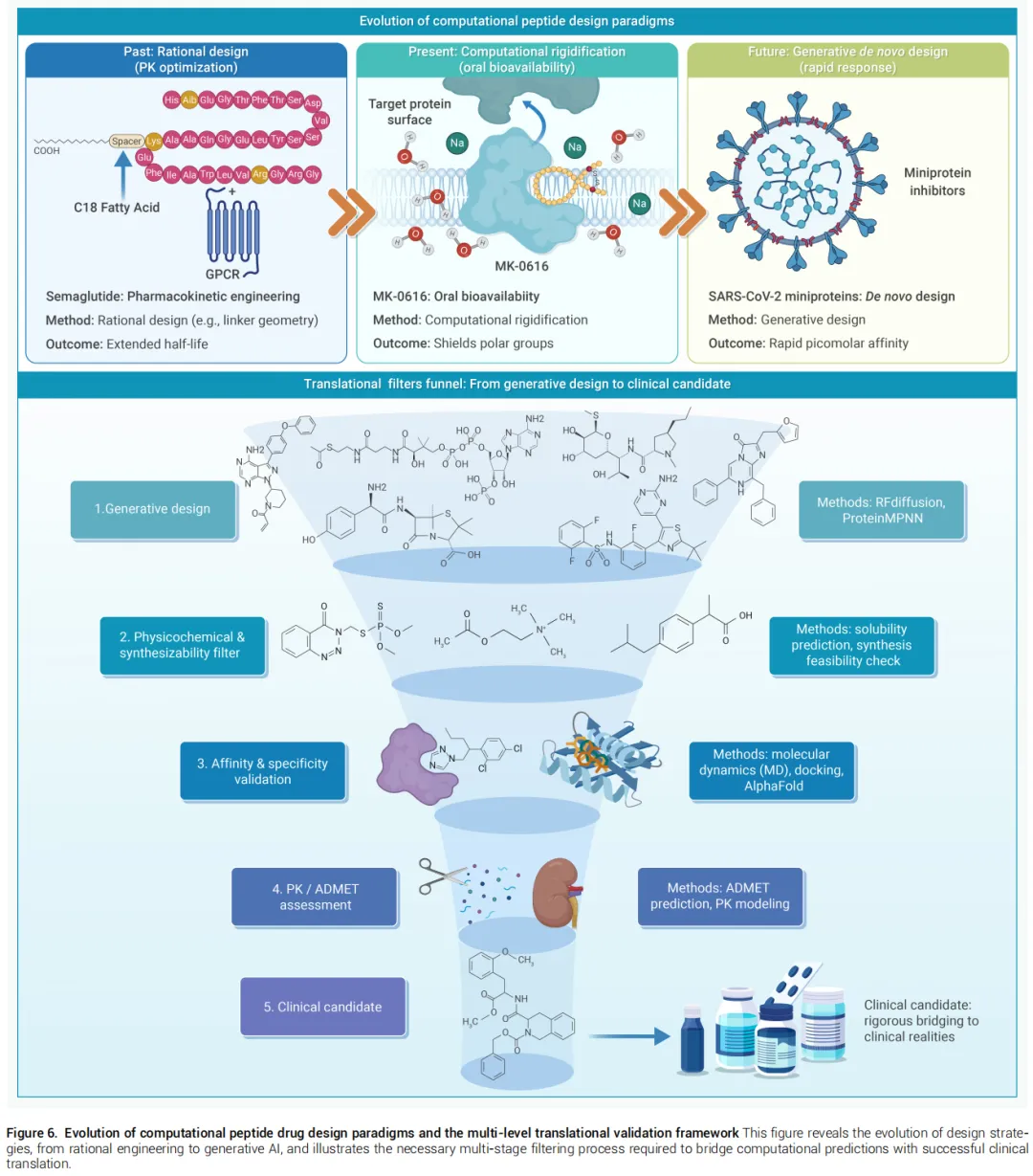
验证层级("validation ladder"):
1. 快速能量最小化/松弛: 立即淘汰Rosetta能量单位严重不利的构象 2. 短时MD模拟(10–50 ns): 以RMSD急剧波动、氢键网络解体为失败判据筛除热力学不稳定设计 3. 自由能计算/湿实验合成: 仅对通过前两层的序列投入高代价资源
非天然氨基酸建模的突破口:当前AI框架的主要瓶颈之一是对非天然氨基酸的支持不足。解决路径包括:扩展模型词汇表并在定制数据集上微调、将残基表示转换为SMILES/SELFIES等化学字符串格式、以及基于三维全原子坐标生成的流匹配/原子扩散框架(如PepFlow),后者从根本上绕过离散标记化限制,无缝整合非典型残基的几何与化学多样性。
六、临床转化案例深度解析
6.1 司美格鲁肽 — 药代动力学的理性工程
问题: 天然GLP-1骨架遭受快速酶解和肾清除,无法维持有效治疗浓度。
计算策略:
• 结构分析精确指导Ala⁸→Aib替换(抗DPP-IV降解) • 计算验证连接臂几何:亲水间隔臂赋予C18脂肪酸二酸足够柔性,实现可逆白蛋白结合(储库效应),同时不干扰GLP-1部分与受体活性位点的关键接触 • MD模拟明确验证连接臂弯曲不影响受体激活
意义: 证明药代动力学性质可作为内在结构特征加以工程化,而非依赖回顾性化学调整。
6.2 MK-0616 — 口服大环多肽的范式转变
问题: 靶向PCSK9的平坦PPI界面需要大尺寸分子,而大分子量与肠道渗透性之间存在根本性矛盾。
计算策略:
• 大环支架的计算辅助刚性化设计将多肽锁定在生物活性构象 • 结构约束最小化结合熵代价,并优化分子内氢键以屏蔽膜转运过程中的极性基团 • 实现了分子量与口服生物利用度的"解耦"
意义: 验证了结构刚性化可解耦分子量与口服生物利用度这一设计范式,为胞内靶点的口服多肽递送开辟路径。
6.3 SARS-CoV-2微蛋白抑制剂 — 计算优先的紧急响应
问题: 新发病原体对快速治疗响应提出极致要求,传统抗体亲和力成熟周期长达数年。
计算策略: 生成算法仅以病毒Spike蛋白几何形状为约束从头设计合成支架,完全绕过漫长的亲和力成熟过程。
结果: 数月内实现皮摩尔级亲和力和动物模型中的强效中和活性。
意义: 标志着"计算优先"发现模式的崛起——高亲和力治疗药物可按需生成,以前所未有的速度和精度应对新兴生物威胁。
七、转化瓶颈与现实挑战
尽管计算设计取得变革性进展,从理论蓝图到临床现实的转化仍面临系统性障碍:
合成可行性鸿沟: 生成模型专注于几何互补性,易引入过度疏水表面或聚集倾向基序,导致化学合成失败或水溶性缺失。
体内药效预测不足: 计算优化常忽视蛋白酶敏感性、肾清除率等关键药代参数,体外高亲和力与体内治疗效力之间存在系统性偏差。
非标准修饰建模局限: 大多数深度生成模型训练于天然氨基酸系统,难以直接处理D-氨基酸、β-氨基酸等非经典修饰,而这些修饰往往是解决代谢稳定性的关键手段。
弥合策略:
• 在生成阶段嵌入显式溶解度约束和负向设计过滤器 • 多目标奖励函数同步优化亲和力、溶解度、蛋白酶稳定性和免疫原性 • 从AI驱动的成药性分析中提取负向设计信号,主动引导生成分布远离有效但不可成药的化学空间
八、未来展望
综述指出了多肽计算设计的三大关键前沿:
8.1 可解释人工智能(XAI)
下一代计算框架需整合可解释AI,阐明驱动预测结果的具体结构特征,从而为构效关系(SAR)提供机制洞见,使计算决策从"黑箱"走向可验证的科学推断。
8.2 多模态数据融合
将一维序列信息、三维几何数据与湿实验功能读数协同整合,构建能够精准导航多肽适应性景观复杂性的鲁棒预测模型,是突破当前预测瓶颈的关键路径。
8.3 化学词汇表的扩展
未来生成框架必须将非经典氨基酸和多样化化学修饰纳入生成过程,使AI模型能够直接优化内在稳定性和生物利用度,从根本上弥合计算命中物与临床候选药物之间的鸿沟。
九、核心结论
| 范式转变 | |
| 最优架构 | |
| 临床差距 | |
| 技术前沿 | |
| 长远愿景 |
