 夜雨聆风
夜雨聆风





精准癌症研究新工具:RESOLVE用更少的突变特征精准解码癌症基因组
癌症基因组的突变特征是解析致癌机制、寻找诊疗靶点的核心线索,但现有分析方法常被冗余特征、过拟合、结果无置信度等问题困扰。本文推出RESOLVE计算框架,通过正则化约束、交叉验证与bootstrap重采样,用更少的核心突变特征实现精准拟合,还能关联患者预后与驱动突变,为癌症研究提供了更可靠的全流程工具。
一、现有突变特征分析的面临的问题
癌症突变特征分析是理解内外源致癌因素、肿瘤演化的关键,但当前研究面临三个核心痛点:
-
特征冗余与过拟合:COSMIC等数据库收录了大量突变特征,其中不少是罕见或过程重叠的冗余特征,分析时易过度拟合样本噪声,难以反映真实致癌过程。 -
缺乏置信度评估:现有方法无法量化突变特征在样本中的活性可靠性,难以区分真实突变过程与随机噪声。 -
临床转化衔接不足:多数分析仅停留在特征提取阶段,缺乏与患者预后、驱动基因突变的精准关联,难以转化为临床可用的生物标志物或机制线索。
二、RESOLVE:少而精的突变特征分析全流程框架
为破解上述问题,研究者开发了RESOLVE计算框架,核心是通过正则化约束+双向交叉验证+bootstrap置信度评估,实现从突变特征提取到临床关联的全流程分析。
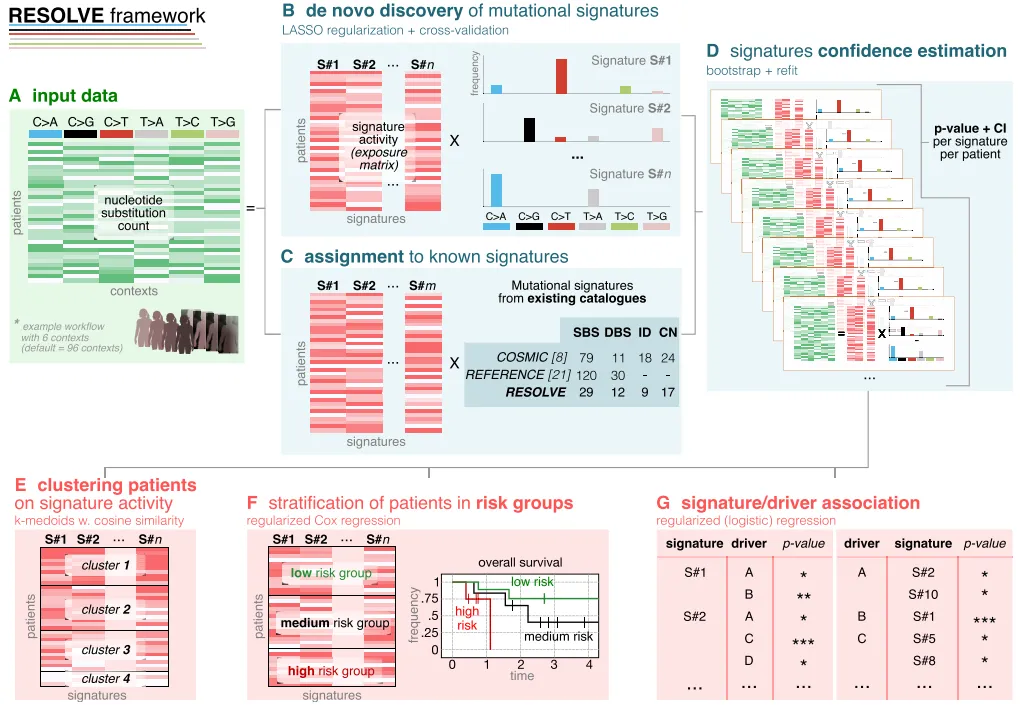 图1:RESOLVE框架的核心步骤,从突变计数输入开始,经正则化特征提取、置信度评估,最终实现患者聚类、预后分析和驱动突变关联。
图1:RESOLVE框架的核心步骤,从突变计数输入开始,经正则化特征提取、置信度评估,最终实现患者聚类、预后分析和驱动突变关联。
具体策略包括:
-
LASSO正则化的非负矩阵分解:同时对突变特征矩阵和样本暴露矩阵施加稀疏性约束,自动筛选对拟合贡献最大的核心特征,减少冗余。 -
双向交叉验证确定最优特征数:通过反复随机掩盖部分数据并评估拟合效果,选择拟合度最高且特征数最少的模型,避免过拟合。 -
bootstrap重采样评估置信度:对样本进行多次重采样后重新分析,统计特征活性分布,给出每个特征在样本中活性的P值,区分真实活性与噪声。 -
整合多模块分析:内置k-medoids聚类、正则化Cox生存分析、正则化回归关联分析模块,直接实现从特征到临床、机制的关联。
三、核心发现:少特征,大信息量
1. 更少的突变特征,媲美全量特征的拟合效果
研究者用RESOLVE对2万多成人+儿科全基因组测序样本提取四类突变特征(SBS、DBS、ID、CN),结果显示:以最常用的SBS特征为例,RESOLVE仅提取29个特征,远少于COSMIC的79个和参考数据库的120个,但拟合效果几乎相当——成人癌症中,RESOLVE的平均余弦相似度为0.984,COSMIC为0.990,参考数据库为0.992,且97.2%的样本拟合度>0.95;儿科癌症中也呈现类似趋势。
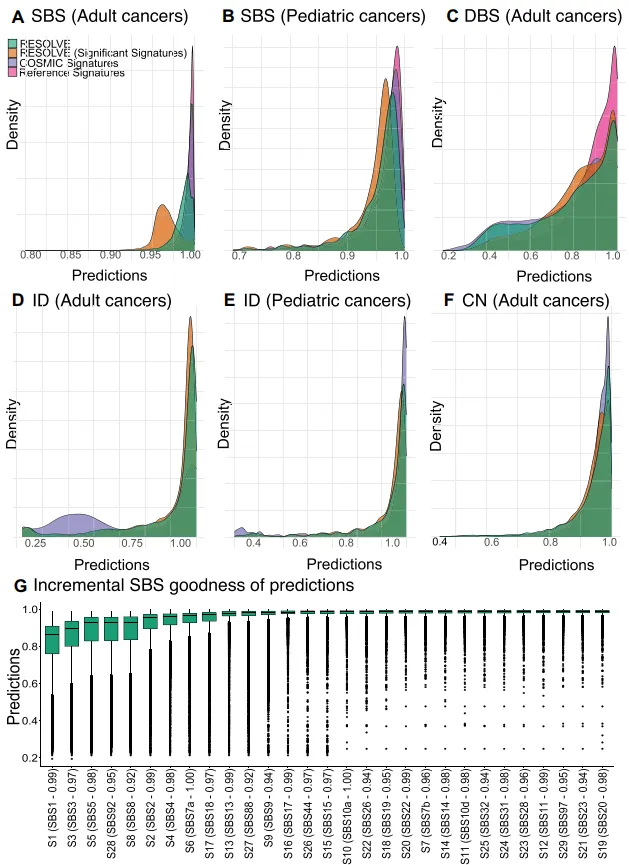 图2:图中展示了SBS、DBS等不同突变类型下,RESOLVE与COSMIC、参考数据库的拟合余弦相似度对比,可见RESOLVE用更少特征实现了接近的拟合精度。
图2:图中展示了SBS、DBS等不同突变类型下,RESOLVE与COSMIC、参考数据库的拟合余弦相似度对比,可见RESOLVE用更少特征实现了接近的拟合精度。
2. 基于突变特征的精准患者聚类
对成人SBS特征的聚类分析发现,患者可分为10个簇,超过60%的患者集中在前3个簇:
-
簇1:主导特征为SBS1(甲基化)+SBS5(衰老) -
簇2:主导特征为SBS3(同源重组缺陷) -
簇3:主导特征为SBS1+SBS13(APOBEC活性)+SBS92(烟草暴露)
儿科癌症的聚类则更简单,仅2个簇,均以甲基化特征(SBS1)为主导。
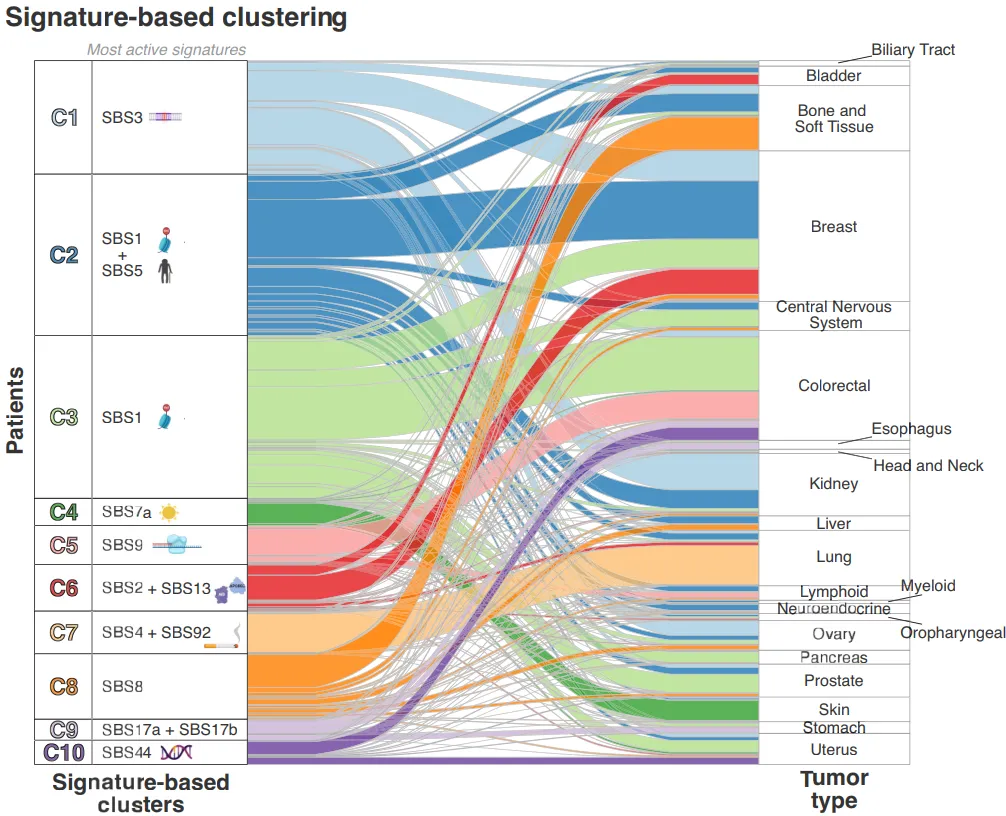 图3:图中展示了不同肿瘤类型在10个簇中的分布,以及每个簇的主导突变特征,可见多数肿瘤类型的患者聚集在少数以核心突变过程为主导的簇中。
图3:图中展示了不同肿瘤类型在10个簇中的分布,以及每个簇的主导突变特征,可见多数肿瘤类型的患者聚集在少数以核心突变过程为主导的簇中。
3. 突变特征与患者预后的明确关联
通过正则化Cox生存分析,研究者发现多个突变特征与预后直接相关:
-
SBS9在造血系统癌症中与更好的预后显著相关; -
SBS3在食管癌、胰腺癌等多种癌症中与更差的预后相关; -
在食管癌症中,低风险组主导特征为SBS5(衰老)+SBS18(ROS损伤),伴随ATM、NTRK3突变,而高风险组则富集KRAS、SMAD4、TP53突变。
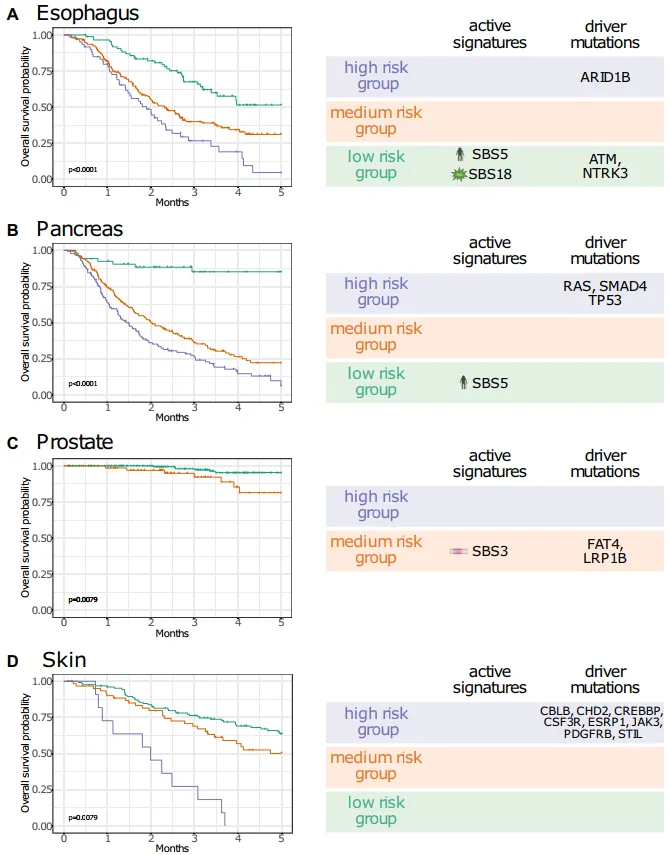 图4:图中展示了食管癌、胰腺癌、前列腺癌和皮肤癌的风险分层生存曲线,以及各风险组对应的活跃突变特征和驱动突变,可见突变特征可有效区分预后差异。
图4:图中展示了食管癌、胰腺癌、前列腺癌和皮肤癌的风险分层生存曲线,以及各风险组对应的活跃突变特征和驱动突变,可见突变特征可有效区分预后差异。
4. 突变特征与驱动基因的双向关联
RESOLVE的双向关联分析揭示了突变特征与驱动基因的复杂关系:
-
特征驱动突变:乳腺癌中SBS2(APOBEC活性)与PIK3CA突变显著关联,推测APOBEC的突变模式直接导致了PIK3CA的特定突变; -
突变影响特征活性:部分驱动突变的突变模式与肿瘤主导特征不符,比如造血系统癌症中EZH2、MYC突变与SBS1、SBS9特征关联,但突变本身并非由这些特征直接导致,提示这些突变可能发生在癌变早期,或受选择压力保护而未被后续突变过程影响。
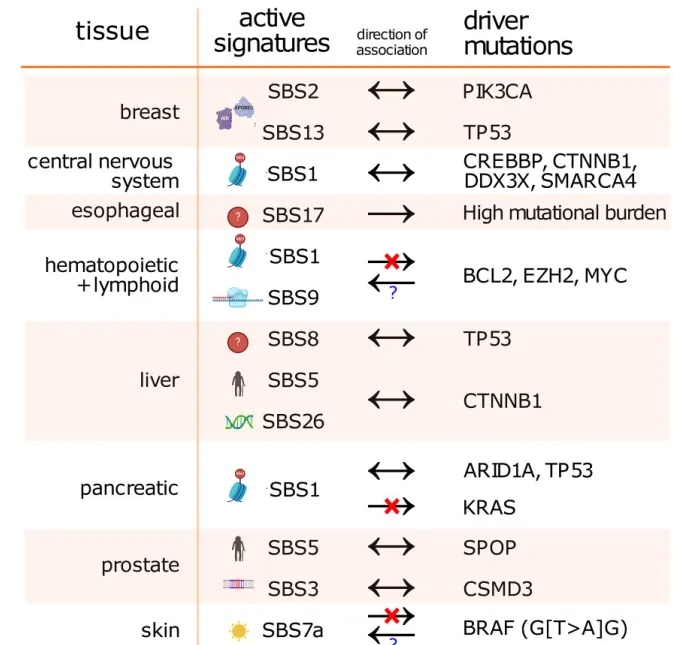 图5:图中展示了不同癌症类型中,核心突变特征与关键驱动基因的关联,箭头表示关联方向,揭示了致癌过程中突变特征与驱动基因的相互作用。
图5:图中展示了不同癌症类型中,核心突变特征与关键驱动基因的关联,箭头表示关联方向,揭示了致癌过程中突变特征与驱动基因的相互作用。
四、RESOLVE的亮点与展望
方法论优势
-
解决过拟合问题:通过LASSO正则化和双向交叉验证,自动筛选核心特征,避免冗余特征带来的过拟合; -
填补置信度评估空白:bootstrap重采样首次为突变特征的活性提供了统计置信度(P值),让结果更可靠; -
全流程整合:从特征提取到临床关联一步到位,无需在多个工具间切换,提升分析效率。
数据与应用优势
-
大样本验证:基于2万多成人+儿科WGS样本验证,覆盖多数癌症类型,结果具有良好的泛化性; -
开源可用:已打包为R包,可从Bioconductor或GitHub获取,方便研究者重复和扩展分析。
临床转化潜力
-
患者分层:基于突变特征的聚类可作为独立的患者分层指标,为精准治疗提供依据; -
预后生物标志物:特定突变特征可作为预后预测的生物标志物,比如SBS3可作为食管癌、胰腺癌的不良预后指标; -
致癌机制解析:突变特征与驱动基因的关联为理解致癌过程提供新线索,比如APOBEC活性与PIK3CA突变的关联,可能为乳腺癌的靶向治疗提供新方向。
局限性与展望
-
当前聚焦主导突变特征,对罕见突变特征的分析不足,未来可优化算法兼顾罕见特征; -
未分析晚期肿瘤或治疗后复发肿瘤的突变特征变化,后续可拓展到动态突变过程研究; -
可整合单细胞基因组数据,分析肿瘤内异质性中的突变特征差异,更深入理解肿瘤演化。
总结
RESOLVE框架通过“少而精”的突变特征分析,打破了“特征越多越精准”的误区,用更少的核心特征实现了媲美全量特征的拟合效果,同时首次为突变特征提供了置信度评估,还直接关联了患者预后与驱动突变。这不仅为癌症突变特征研究提供了更可靠的工具,也为精准癌症诊疗的生物标志物开发和机制研究开辟了新方向。随着更多样本和数据类型的整合,RESOLVE有望成为癌症基因组研究的标准工具之一。
原文信息
-
Comprehensive analysis of mutational processes across 20 000 adult and pediatric tumors. Nucleic Acids Res. 2025 Jul 10;53(13):gkaf648. -
https://doi.org/10.1093/nar/gkaf648
都看到了这里,辛苦关注一下吧!☟☟☟