 夜雨聆风
夜雨聆风





如何选择GWAS分析软件
面对不同的样品状况,特别是大规模样品库,首先要考虑哪一款GWAS软件适合自己的数据。本文比较了BOLT-LMM、SAIGE、REGENIE、PLINK 2.0、GCTA-fastGWA这5款软件的优势,缺点和适用场景。
下面是推荐的决策树,详细的比较信息可以继续阅读。
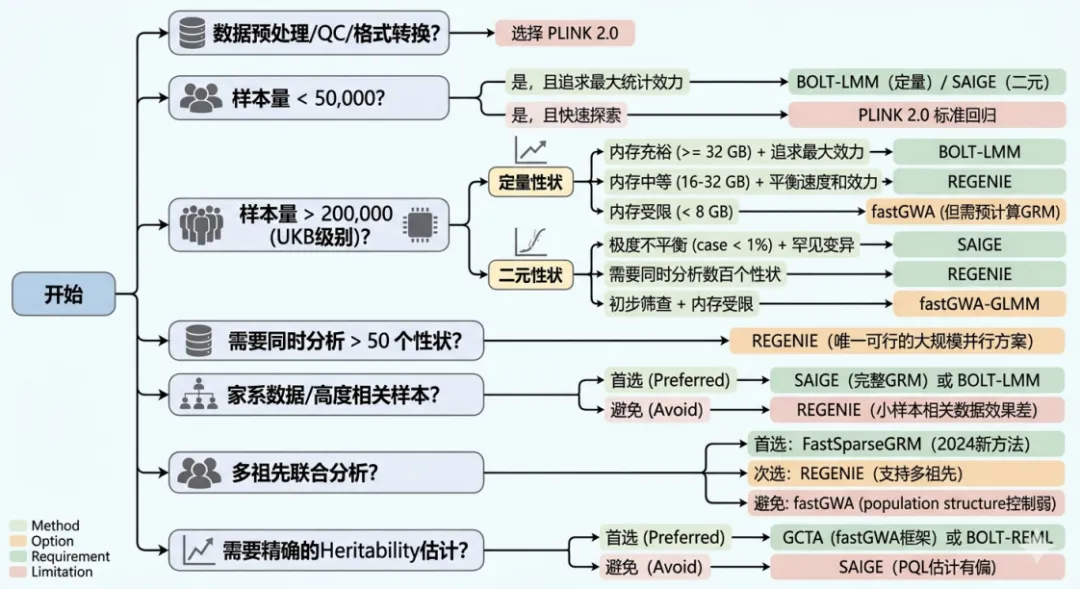
一、总体比较概览
|
软件 |
第一作者/团队 |
发表时间 |
核心算法 |
最大测试样本量 |
适用性状类型 |
内存需求 |
核心定位 |
|
BOLT-LMM |
Loh et al. (Broad Institute) |
2015, Nature Genetics |
非无穷小LMM + PCG迭代 |
~500,000 (UKB) |
定量/平衡二元 |
~50 GB |
统计效力最高的LMM |
|
SAIGE |
Zhou et al. (University of Michigan) |
2018, Nature Genetics |
PQL + SPA校正 |
~500,000 (UKB) |
不平衡二元/罕见变异 |
<15 GB |
不平衡二元性状专家 |
|
REGENIE |
Mbatchou et al. (Regeneron) |
2021, Nature Genetics |
全基因组岭回归 + LOCO |
~500,000 (UKB) |
定量/二元/海量性状 |
~13 GB |
超大规模速度之王 |
|
PLINK 2.0 |
Chang et al. (Harvard/Stanford) |
2015-持续更新 |
多元回归 + 多样方法 |
无上限 |
全类型 |
可变 |
通用GWAS平台 |
|
GCTA-fastGWA |
Jiang et al. (Westlake University) |
2019/2021, Nature Genetics |
稀疏GRM + REML |
~500,000 (UKB) |
定量/二元 |
~2 GB |
内存极致优化 |
二、BOLT-LMM
2.1 核心信息
|
项目 |
内容 |
|
参考文献 |
Loh PR, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics. 2015;47:284-290. |
|
算法基础 |
线性混合模型(LMM),假设SNP效应服从正态分布,但允许非无穷小(non-infinitesimal)遗传架构 |
|
核心算法 |
预条件共轭梯度法(PCG)求解线性系统;2-bit压缩存储原始基因型;利用Level 3 BLAS和SSE指令加速 |
|
实现语言 |
C++ |
|
最新版本 |
BOLT-LMM v2.4 |
2.2 核心优势
|
优势 |
具体说明 |
|
统计效力最高 |
非无穷小模型(Bayesian spike-slab prior)对真实遗传架构的拟合优于无穷小假设,在模拟和实际数据中均显示最强关联信号 |
|
P值校准优秀 |
对定量性状和相对平衡的二元性状(case:control ≈ 1:1至1:10),I类错误控制严格,P值无膨胀 |
|
无需预计算GRM |
直接操作原始基因型文件(2-bit压缩),无需存储N×N的稠密遗传关系矩阵(GRM) |
|
共轭梯度加速 |
PCG迭代求解替代直接矩阵求逆,Step 1计算复杂度从O(N³)降至O(N¹·⁵M) |
|
多线程优化 |
支持多线程并行,充分利用现代CPU的SIMD指令集 |
2.3 主要缺点
|
缺点 |
具体说明 |
|
仅限定量/平衡二元性状 |
原生设计针对定量性状。对不平衡二元性状(case:control < 1:10),传统Score检验P值严重膨胀 |
|
计算速度极慢 |
在UK Biobank规模(N~400,000,50个表型)下,Step 1(null model fitting)需要~1,150 CPU小时,Step 2需要~7,250 CPU小时,总计~8,400 CPU小时(约350天单核) |
|
内存需求大 |
Step 1需要约50 GB RAM,主要由于需在内存中保存部分基因型矩阵和迭代中间结果 |
|
不支持罕见变异 |
传统BOLT-LMM未集成SPA或Firth校正,对MAF<0.5%的罕见变异检验效力不足 |
|
成本高昂 |
在UKB RAP平台上分析50个定量性状的成本约£7,500(约7万元人民币),是REGENIE的310倍 |
|
不纳入PCs时I类错误膨胀 |
若不将主成分(PCs)作为固定效应纳入模型,可能出现population structure导致的假阳性 |
2.4 适用场景
•首选场景:
–数量性状(身高、BMI、血脂等连续表型)的GWAS
–相对平衡的二元性状(如冠心病,case:control ≈ 1:10)
–样本量中等(N < 100,000)且追求最大统计效力的研究
–遗传力较高(h² > 0.2)且遗传架构可能非无穷小的性状
•不推荐场景:
–极度不平衡的二元性状(如罕见病,case:control < 1:50)
–内存受限的计算环境(< 32 GB RAM)
–需要同时分析数百个性状的大规模项目
三、SAIGE
3.1 核心信息
|
项目 |
内容 |
|
参考文献 |
Zhou W, Nielsen JB, Fritsche LG, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics. 2018;50:1335-1341. |
|
算法基础 |
广义线性混合模型(GLMM)+ 惩罚拟似然(PQL)+ SPA(Saddle Point Approximation)校正 |
|
核心算法 |
PQL估计随机效应方差;Score检验 + SPA校正处理不平衡和罕见变异;稀疏GRM |
|
实现语言 |
C++ / R接口 |
|
最新版本 |
SAIGE-GENE / SAIGE+(支持区域检验) |
3.2 核心优势
|
优势 |
具体说明 |
|
不平衡二元性状专家 |
专为case-control不平衡设计(如甲状腺癌 case:control = 1:660),SPA校正使P值在极端不平衡下仍严格校准 |
|
罕见变异友好 |
唯一能在UKB规模(N~400,000)中可靠检测MAF < 0.5%罕见变异的方法。原始论文发现1,609个MAF<0.5%的显著变异 |
|
I类错误控制严格 |
在模拟中,即使case:control = 1:1,000和MAF = 0.01%,P值分布仍保持均匀 |
|
样本相关性处理 |
通过稀疏GRM有效处理家系数据和隐性亲属关系 |
|
OR估计 |
提供可靠的效应量(odds ratio)估计,优于纯Score检验方法 |
|
广泛使用 |
全球大型生物库(UKB、TOPMed、MVP)的首选二元性状分析工具 |
3.3 主要缺点
|
缺点 |
具体说明 |
|
计算极慢 |
Step 1(null model fitting)在N~400,000时需要~9 CPU小时;Step 2需要~12,015 CPU小时(50个二元性状),总计~12,024 CPU小时(约500天单核),是REGENIE的14倍 |
|
收敛不稳定 |
PQL算法的收敛时间因表型遗传力、样本相关性和case-control比例而异;某些表型可能无法收敛 |
|
极度不平衡时略保守 |
当case比例<1%时,P值分布可能略微保守(假阴性率增加) |
|
遗传力估计有偏 |
PQL估计的方差组分τ²有偏,不能用于heritability估计 |
|
假设无穷小架构 |
假设SNP效应服从正态分布,对非无穷小遗传架构(如少数大效应位点)可能损失统计效力 |
|
OR计算不高效 |
准确OR估计需要拟合备择模型,计算成本高;当前版本使用null model参数近似估计OR |
|
成本极高 |
UKB RAP平台上50个二元性状分析成本约£2,835(约2.6万元人民币),是fastGWA-GLMM的29倍 |
|
定量性状支持弱 |
SAIGE主要设计用于二元性状,定量性状分析非其专长 |
3.4 适用场景
•首选场景:
–不平衡二元性状(罕见病、癌症亚型、甲状腺疾病等case比例<5%)
–罕见变异(MAF < 1%)的GWAS
–家系数据或存在隐性亲属关系的队列
–对P值校准要求极高的研究(如药物靶点验证)
•不推荐场景:
–同时分析大量(>50个)二元性状(计算成本不可承受)
–需要精确的SNP-based heritability估计
–遗传架构明显非无穷小(如主效基因)
四、REGENIE
4.1 核心信息
|
项目 |
内容 |
|
参考文献 |
Mbatchou J, Barnard L, Backman J, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nature Genetics. 2021;53:1097-1103. |
|
算法基础 |
全基因组岭回归(Ridge Regression)+ Leave-One-Chromosome-Out (LOCO)残差 |
|
核心算法 |
Step 1: 分块岭回归估计多基因效应(SNP按LD block分块);Step 2: 用LOCO残差进行边际SNP检验 |
|
实现语言 |
C++ |
|
最新版本 |
REGENIE v4.x(支持定量/二元/生存/Ordinal性状,以及quantile regression Regenie.QRS) |
4.2 核心优势
|
优势 |
具体说明 |
|
速度之王 |
Step 1比BOLT-LMM快150倍,比SAIGE快300倍。UKB 50个定量性状总计仅需~50 CPU小时(约2天),成本仅£24 |
|
海量性状并行 |
天然支持多表型同时分析(Multi-trait),100个表型可一次性完成Step 1 |
|
内存友好 |
最大内存使用~13 GB(vs BOLT-LMM 50 GB),通过分块读取基因型数据实现 |
|
Firth/SPA双重校正 |
对不平衡二元性状提供Firth惩罚似然和SPA两种校正,P值校准与SAIGE相当 |
|
LOCO策略 |
Leave-One-Chromosome-Out避免近端污染(proximal contamination),提高检验准确性 |
|
支持超大规模 |
已验证可在UKB全队列(N500,000,1,160万SNP)上稳定运行 |
|
多祖先支持 |
2023年后版本支持多祖先生物库的联合分析 |
4.3 主要缺点
|
缺点 |
具体说明 |
|
仅支持无穷小模型 |
岭回归本质上是LMM的无穷小近似(ridge penalty ≈ 1/τ²),对非无穷小遗传架构的统计效力低于BOLT-LMM(模拟中差距5-10%) |
|
小样本相关数据表现差 |
在小型家系数据(N < 10,000)中,由于稀疏GRM近似的局限性,P值可能不够准确 |
|
磁盘空间需求大 |
Step 1需要约355 GB磁盘空间存储中间文件(LOCO预测值),对存储系统压力大 |
|
定量性状效力略低 |
在已知峰值的关联分析中,REGENIE的信号强度略弱于BOLT-LMM(差约5-10%) |
|
不支持非标准模型 |
不支持基因×环境交互、非线性效应等复杂模型 |
|
稀疏GRM效力限制 |
与fastGWA类似,稀疏GRM近似在多祖先数据中可能损失部分统计效力 |
4.4 适用场景
•首选场景:
–UK Biobank/TOPMed级别超大规模生物库(N > 200,000)
–需要同时分析数百个性状的项目(如UKB 1,500+表型全扫描)
–内存受限但存储充裕的计算环境
–不平衡二元性状的大规模筛查(如所有ICD编码疾病)
•不推荐场景:
–小型家系数据(N < 10,000,高度相关个体)
–追求单个性状最大统计效力的精细映射研究
–存储空间极度受限的环境
五、PLINK 2.0
5.1 核心信息
|
项目 |
内容 |
|
参考文献 |
Chang CC, Chow CC, Tellier LC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. |
|
算法基础 |
多元线性/逻辑回归、Firth惩罚似然、SKAT-O等多样方法集合 |
|
核心定位 |
通用GWAS数据处理和分析平台,非专门的LMM工具 |
|
实现语言 |
C |
|
最新版本 |
PLINK 2.00 alpha(持续更新) |
5.2 核心优势
|
优势 |
具体说明 |
|
最全面的QC功能 |
数据质控、过滤、格式转换、LD计算、PCA、亲缘关系推断等功能一应俱全,是GWAS pipeline的标准起点 |
|
文件格式兼容性 |
支持VCF/BCF/BGEN/PGEN/BED+二进制格式,与所有主流LMM工具(BOLT-LMM/SAIGE/REGENIE)无缝衔接 |
|
多种关联分析方法 |
线性回归、逻辑回归、Firth回归、EMMAX近似、SKAT/SKAT-O区域检验、set-based检验等 |
|
成熟的生态系统 |
用户群体最大,文档最完善,社区支持最强,教程和最佳实践资源极其丰富 |
|
PGEN格式 |
专有的高性能基因型存储格式(2-bit + variant-differential encoding),读写速度远超传统BED格式 |
|
命令行友好 |
简洁直观的命令行接口,易于整合到自动化pipeline |
5.3 主要缺点
|
缺点 |
具体说明 |
|
无内置LMM/GRM |
PLINK 2.0本身不是LMM工具,对population structure和样本相关性的校正能力有限;必须配合BOLT-LMM/SAIGE/REGENIE等外部工具 |
|
大规模数据效率不足 |
在N > 100,000时,标准回归分析的速度和内存效率不如专用LMM工具 |
|
二元性状默认无SPA |
逻辑回归对不平衡性状和罕见变异的P值膨胀问题未自动校正 |
|
无LOCO策略 |
不提供leave-one-chromosome-out功能,近端污染可能导致效应估计偏倚 |
|
不支持Bayesian模型 |
无非无穷小或Bayesian稀疏先验模型 |
5.4 适用场景
•首选场景:
–GWAS数据质控和预处理(QC、过滤、格式转换、PCA计算)
–小型到中型队列(N < 50,000)的标准回归分析
–与BOLT-LMM/REGENIE/SAIGE配合的完整pipeline(PLINK做QC → LMM工具做关联)
–候选基因分析、基因–环境交互、非线性模型探索
–教育和培训(最直观的GWAS入门工具)
•不推荐场景:
–作为大型生物库(N > 200,000)的主要关联分析工具
–高度不平衡的二元性状分析
–需要LOCO校正的全基因组精细映射
六、GCTA-fastGWA
6.1 核心信息
|
项目 |
内容 |
|
参考文献 |
Jiang L, Zheng Z, Qi T, et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nature Genetics. 2019;51:1749-1755. (定量) Jiang L, Zheng Z, Tang W, et al. Methods for constructing polygenic risk scores based on LASSO and best linear unbiased prediction. 2021 (二元, fastGWA-GLMM) |
|
算法基础 |
稀疏GRM近似+ REML/PQL方差组分估计 |
|
核心算法 |
利用GRM的稀疏性(仅保留近亲关系对),将N×N稠密矩阵转为稀疏矩阵,大幅降低计算和内存需求 |
|
实现语言 |
C++ / R (GCTA框架) |
|
最新版本 |
fastGWA / fastGWA-GLMM (GCTA v1.94+) |
6.2 核心优势
|
优势 |
具体说明 |
|
内存极致优化 |
最大内存仅~2 GB(vs BOLT-LMM 50 GB,REGENIE 13 GB),是五款工具中内存需求最低的 |
|
计算效率高 |
Step 2(全基因组扫描)速度快,UKB 50个定量性状约128 CPU小时(vs BOLT-LMM 7,250小时) |
|
稀疏GRM一次性构建 |
GRM只需计算一次,后续可重复用于多个表型 |
|
成本较低 |
UKB RAP平台50个定量性状成本£37,50个二元性状£98,是BOLT-LMM的1/200 |
|
基于成熟框架 |
建立在GCTA/GREML的严格统计理论基础上,heritability估计可靠 |
|
定量/二元双支持 |
fastGWA(定量)和fastGWA-GLMM(二元)覆盖两种主要表型 |
6.3 主要缺点
|
缺点 |
具体说明 |
|
统计效力最低 |
稀疏GRM近似损失了LMM的polygenic校正能力。在模拟中,fastGWA的empirical power显著低于BOLT-LMM和REGENIE(尤其当variant与PCs相关时,power趋近于零) |
|
Population structure控制不足 |
稀疏GRM主要捕获近亲关系,对远缘population structure的校正能力弱于完整LMM。在多祖先数据中,未校正population structure可导致大量假阳性 |
|
Family data过于保守 |
在家系数据中,fastGWA-GLMM的P值分布过于保守(大量真信号被遗漏) |
|
GRM构建成本高 |
虽然每次分析内存低,但构建稀疏GRM的一次性成本不可忽视:UKB 368,000人需要763 CPU小时 |
|
二元性状需降阈 |
由于包含罕见变异增加多重检验负担,fastGWA-GLMM建议使用P < 5×10⁻⁹(而非标准5×10⁻⁸)作为显著性阈值 |
|
方差组分不可解释 |
fastGWA-GLMM估计的σ²θ不能解释为遗传方差或heritability(PQL和Laplace近似导致) |
|
GCTA-LOCO不可行 |
传统的GCTA-LOCA分析在UKB规模完全不可行(估计需要>500 GB内存和>10,000 CPU小时) |
6.4 适用场景
•首选场景:
–内存极度受限的计算环境(如共享集群的 limited memory节点)
–已知GRM已预计算的情况下,快速扫描大量表型
–同质性人群(单祖先)的初步GWAS筛查,population structure已通过PCs充分校正
–遗传力较低(h² < 0.1)的性状,polygenic效应较弱
•不推荐场景:
–多祖先联合分析(population structure复杂)
–家系数据或高度相关的样本
–追求最大统计效力的精细映射研究
–需要同时校正population structure和polygenic effect的研究
七、五款软件横向深度对比
7.1 计算性能对比(UK Biobank规模,N ≈ 400,000)
|
软件 |
Step 1时间 |
Step 2时间 |
总时间(50表型) |
最大内存 |
RAP成本(£) |
|
BOLT-LMM |
~1,150 h |
~7,250 h |
~8,400 h |
~50 GB |
~7,500 |
|
SAIGE |
~9 h |
~12,015 h |
~12,024 h |
<15 GB |
~2,835 |
|
REGENIE |
~4.7 h |
~45.3 h |
~50 h |
~13 GB |
~24 |
|
fastGWA |
763 h(GRM) |
~128 h |
~891 h |
~2 GB |
~37 |
|
fastGWA-GLMM |
763 h(GRM) |
~254 h |
~1,017 h |
~2 GB |
~98 |
|
PLINK 2.0 |
N/A |
N/A |
不适用 |
可变 |
不适用 |
数据来源:Quickdraws 2024论文中的UKB RAP实际运行数据(Nature Genetics, 2024)
7.2 统计效力对比(模拟研究)
|
软件 |
定量性状power |
二元性状power |
罕见变异(MAF<0.5%) |
多祖先数据 |
|
BOLT-LMM |
★★★★★ (最高) |
★★★☆☆ (平衡时) |
★★☆☆☆ |
★★★☆☆ |
|
SAIGE |
★★☆☆☆ |
★★★★★ (不平衡) |
★★★★★ |
★★★★☆ |
|
REGENIE |
★★★★☆ (仅次于BOLT) |
★★★★☆ |
★★★☆☆ |
★★★★☆ |
|
fastGWA |
★★☆☆☆ |
★★☆☆☆ |
★★☆☆☆ |
★☆☆☆☆ |
|
PLINK 2.0 |
★★☆☆☆ (标准回归) |
★★☆☆☆ |
★☆☆☆☆ |
★★☆☆☆ |
7.3 核心功能矩阵
|
功能 |
BOLT-LMM |
SAIGE |
REGENIE |
PLINK 2.0 |
fastGWA |
|
定量性状 |
✓✓✓ |
✓ |
✓✓✓ |
✓✓ |
✓ |
|
平衡二元性状 |
✓✓ |
✓✓✓ |
✓✓✓ |
✓ |
✓✓ |
|
不平衡二元性状 |
✗ |
✓✓✓ |
✓✓✓ |
✓ (Firth) |
✓✓ |
|
罕见变异(MAF<0.5%) |
✗ |
✓✓✓ |
✓✓ |
✓ |
✗ |
|
LOCO校正 |
✓ |
✓ |
✓✓✓ |
✗ |
✗ |
|
SPA校正 |
✗ |
✓✓✓ |
✓✓ |
✗ |
✓ |
|
Firth校正 |
✗ |
✗ |
✓✓✓ |
✓✓ |
✗ |
|
Heritability估计 |
✗ |
✗ (有偏) |
✗ |
✗ |
✓✓ (GCTA) |
|
多性状并行 |
✗ |
✗ |
✓✓✓ |
✗ |
✗ |
|
基因×环境 |
✗ |
✗ |
✗ |
✓✓ |
✗ |
|
区域检验(SKAT) |
✗ |
✓ (SAIGE-GENE) |
✗ |
✓✓✓ |
✗ |
|
数据QC/格式转换 |
✗ |
✗ |
✗ |
✓✓✓ |
✗ |
八、新兴工具展望
|
工具 |
年份 |
核心创新 |
状态 |
|
FastSparseGRM |
2024 |
BDSA-GRM + 分区population structure控制 |
预印 |
|
LDAK-KVIK |
2024 |
变分推断加速,定量和二元双支持 |
预印 |
|
Quickdraws |
2024 |
GPU加速贝叶斯回归,>30×速度提升 |
Nature Genetics |
|
SAIGE-GENE |
2023 |
SAIGE扩展至基因/区域水平罕见变异burden检验 |
已发布 |
|
Regenie.QRS |
2025 |
分位数回归GWAS,检测异质性遗传效应 |
预印 |
九、参考文献
1.BOLT-LMM: Loh PR, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics. 2015;47:284-290.
2.BOLT-LMM v2.3: Loh PR, et al. Mixed-model association for biobank-scale datasets. Nature Genetics. 2018;50:906-908.
3.SAIGE: Zhou W, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics. 2018;50:1335-1341.
4.REGENIE: Mbatchou J, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nature Genetics. 2021;53:1097-1103.
5.fastGWA: Jiang L, et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nature Genetics. 2019;51:1749-1755.
6.fastGWA-GLMM: Jiang L, et al. fastGWA-GLMM: a generalized linear mixed model association tool for biobank-scale data. 2021.
7.PLINK 2.0: Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7.
8.Gurinovich A, et al. Evaluation of GENESIS, SAIGE, REGENIE and fastGWA-GLMM for genome-wide association studies of binary traits in correlated data. Frontiers in Genetics. 2022;13:897210.(方法学比较)
9.Powell JF, et al. A scalable variational inference approach for increased mixed-model association power. Nature Genetics. 2024. (Quickdraws)
10.FastSparseGRM: Lin, et al. Scalable analysis of large multi-ancestry biobanks byleveraging sparse ancestry-adjusted sample-relatedness. Res Sq. 2024.