 夜雨聆风
夜雨聆风





AI for Science,大连理工大学,Advanced Science

当 AI for Science 进入新能源材料发现,一个最直接的问题就是:能不能把过去“算不起、筛不动”的巨大化学空间,真正变成可以高效搜索的材料宝库?
近日,大连理工大学团队在 Advanced Science 发表研究,提出了一套面向有机单线态裂分分子的高通量筛选框架,将图神经网络与多层级物理验证结合起来,用于加速发现下一代高效光伏材料候选分子。
这项工作瞄准的是一个非常重要的方向:单线态裂分。
所谓单线态裂分,是指一个高能单线态激子可以分裂成两个低能三线态激子。简单来说,就是一个高能光子原本只能贡献一个激子,而通过单线态裂分,有机会变成两个可利用的激子。这意味着它有潜力突破传统太阳能电池的 Shockley–Queisser 极限,为更高效率、更可持续的光伏器件提供新路径。
但问题也很现实:真正好用的单线态裂分材料并不多。
一个分子要成为理想候选,不仅要满足严格的激发态能级条件,还要具有足够强的可见光吸收、合适的三线态能量、良好的稳定性和可合成性。传统方法通常依赖 TDDFT、GW+BSE 等量子化学计算来评估激发态性质,但这些方法计算成本很高。一旦面对数百万、上千万个候选分子,逐个计算几乎不现实。
这正是 AI 能发挥作用的地方。
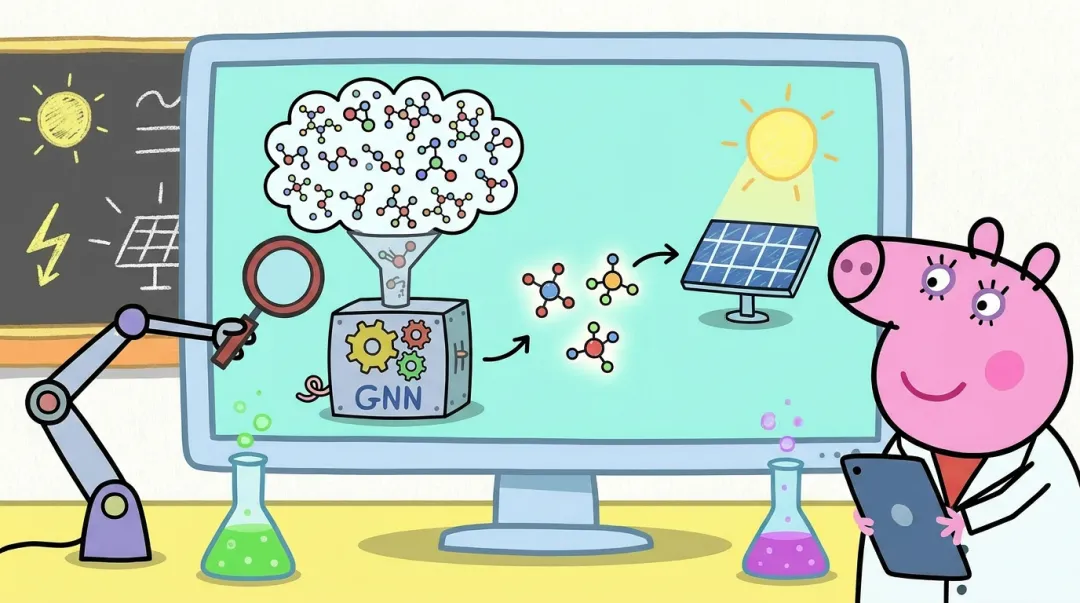
研究团队构建的图神经网络模型,把分子结构转化为分子图:原子是节点,原子间距离和相互作用是边,模型通过多层图卷积不断聚合局部化学环境信息,从而预测与单线态裂分密切相关的激发态性质,包括 S1、T1 和 T2 能级。
这套模型最关键的优势,是在速度和精度之间取得了非常好的平衡。模型在 FORMED 数据库上训练,能够以约 0.1 eV 的平均绝对误差预测关键激发态能量。相比已有机器学习模型,这一精度有明显提升,也为后续大规模筛选提供了可靠基础。
更重要的是,它不仅“会预测”,还真的拿去筛了。
研究团队利用训练好的 GNN 模型,对来自 OE62 和 QO2Mol 数据库的超过 2000 万个有机分子结构进行了快速预测。这个规模如果完全依赖传统 TDDFT 计算,计算量会极其庞大;而借助机器学习,团队先用模型快速缩小候选范围,再对筛出的分子进行 TDDFT 验证,从而将需要进一步量子化学计算的任务从 2000 多万级别压缩到约 5000 个,计算需求降低了几个数量级。
这就是 AI for Science 最打动人的地方:不是简单替代第一性原理计算,而是把昂贵计算用在最值得算的地方。
在这套筛选流程中,AI 负责快速探索巨大化学空间,TDDFT 负责中层验证,GW+BSE 则用于更高精度的最终确认。这样的多层级策略,既避免了纯机器学习可能带来的误判,也避免了纯量子化学筛选无法承受的计算成本。
最终,研究团队筛选出 180 个具有单线态裂分潜力的有机分子,以及超过 1000 个相关构象。这些候选分子不仅在能级条件上满足要求,还展现出多样的结构类型,包括稠环芳香体系、杂原子掺杂结构和功能化有机骨架等,为后续实验合成和器件验证提供了新的分子库。
更值得注意的是,团队没有止步于“算出一批候选”。他们进一步引入合成可及性评估,对候选分子进行实验可行性筛选。对于功能材料发现来说,这一步非常重要。因为一个分子在计算上再漂亮,如果难以合成、稳定性差、成本过高,也很难真正走向应用。
研究中,团队使用 DeepSA 模型评估分子的可合成性,并从 180 个候选分子中筛出 79 个较易合成的分子。随后,又对这些分子开展 GW+BSE 计算进行更高精度验证,最终得到一批同时兼顾单线态裂分潜力和实验可行性的优质候选。
这篇文章的亮点,也正在于它把“AI 预测”推进成了一条完整的材料发现流水线:从数据库出发,经过 GNN 快速预测、能级条件筛选、TDDFT 验证、合成可及性评估,再到 GW+BSE 高精度确认,形成了一个层层收敛、逐级提纯的智能筛选框架。
这不是单点模型的胜利,而是科研流程的升级。
过去,寻找单线态裂分材料很像在巨大化学空间里“摸索”。研究者知道大方向,但每一步都要付出高昂计算和实验成本。现在,这项工作展示了一种更高效的路径:让 AI 先在千万级分子空间里快速扫描,把真正有希望的区域标出来,再用高精度理论方法逐步验证。
这样的范式不仅适用于单线态裂分材料,也可以迁移到更多由激发态性质决定功能的分子体系中,例如荧光材料、磷光材料、三重态—三重态湮灭材料、热活化延迟荧光材料等。
从更大的角度看,这篇 Advanced Science 的意义,在于它回答了 AI for Science 在功能分子发现中的一个核心问题:面对几乎无限的化学空间,AI 能不能帮助我们更快、更准、更系统地找到真正值得实验验证的候选?
这项工作给出的答案非常清晰:可以。
它让人看到,未来的材料发现不再只是“靠经验猜结构”,也不再只是“靠算力硬扫库”,而是通过 AI 和物理计算的协同,把化学空间变成可以被高效导航的地图。
AI for Science,大连理工大学,Advanced Science。
这项工作不只是筛出了一批新的单线态裂分候选分子,更重要的是,它展示了一种面向可持续光伏材料发现的新范式:用图神经网络打开巨大分子空间,用多层级计算验证守住科学可靠性,用合成可及性评估连接实验落地。
当 AI 能够在 2000 万个分子中快速找到真正有潜力的光伏材料候选,功能材料发现的速度,正在被重新定义。
本文不含AI生成内容
欢迎留言分享你的看法