 夜雨聆风
夜雨聆风





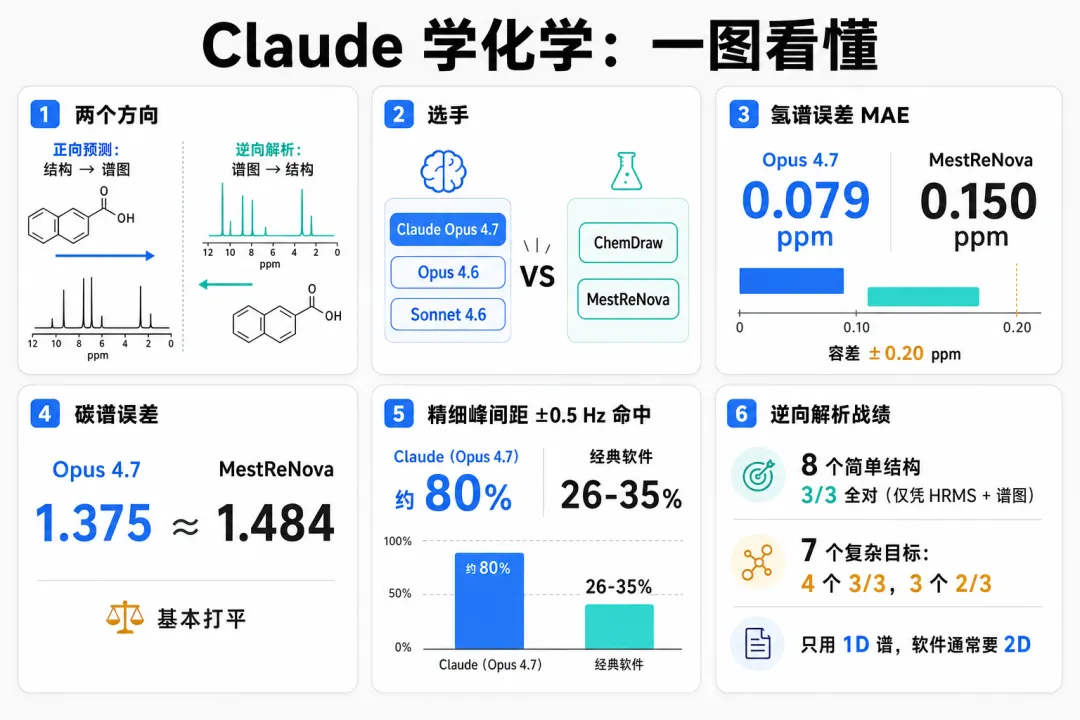
AI for Science 爆发:AI 化学突破进化,Claude 击败化学软件 ChemDraw

Anthropic 用 20 个 ChemRxiv 预印本里的化合物做了一次盲测:在 NMR 化学位移、多重性、耦合常数三项上,Opus 4.7 全面追平或反超 ChemDraw 25.0.2 和 MestReNova 17.0.0,还从一维谱 + 质谱反推出了结构——后者是专用软件做不了的事。
Anthropic 把 Claude 送进了化学实验室
6 月 5 日,Anthropic 内部化学家 David Kamber 发布报告《How Claude performs on NMR prediction and structure elucidation》。
测试对象是三个 Claude 模型:Opus 4.7、Opus 4.6、Sonnet 4.6。对手是两款摆在几乎每个化学家桌面上的专业软件:ChemDraw 25.0.2 和 MestReNova 17.0.0。
战场选在 NMR(核磁共振)谱图。合成一个化合物后,确认手里做出来的是不是目标分子,靠 NMR。把谱图里每个峰对应到结构里每个原子,是合成化学最耗时的环节之一。
让 Claude 接管谱图分析,是 Anthropic AI for Science 计划扩到化学的第一份公开工作。
读一张 NMR 谱:三层信息,两个方向
一张 NMR 谱图是一列峰,每个化学上不等价的氢或碳一个峰,峰的位置被周围原子推移。看懂它要同时读三层信息。
化学位移(chemical shift,单位 ppm):峰在谱图上的位置,反映原子所处的化学环境。预测氢谱和碳谱位移,是最基础的一关。
多重性(multiplicity):一个氢信号的裂分形状——单峰、双峰、三重峰等,由相邻氢的数目决定。
耦合常数(J-coupling,单位 Hz):裂分出来的子峰之间的间距,衡量两个核通过化学键相互作用的强度。
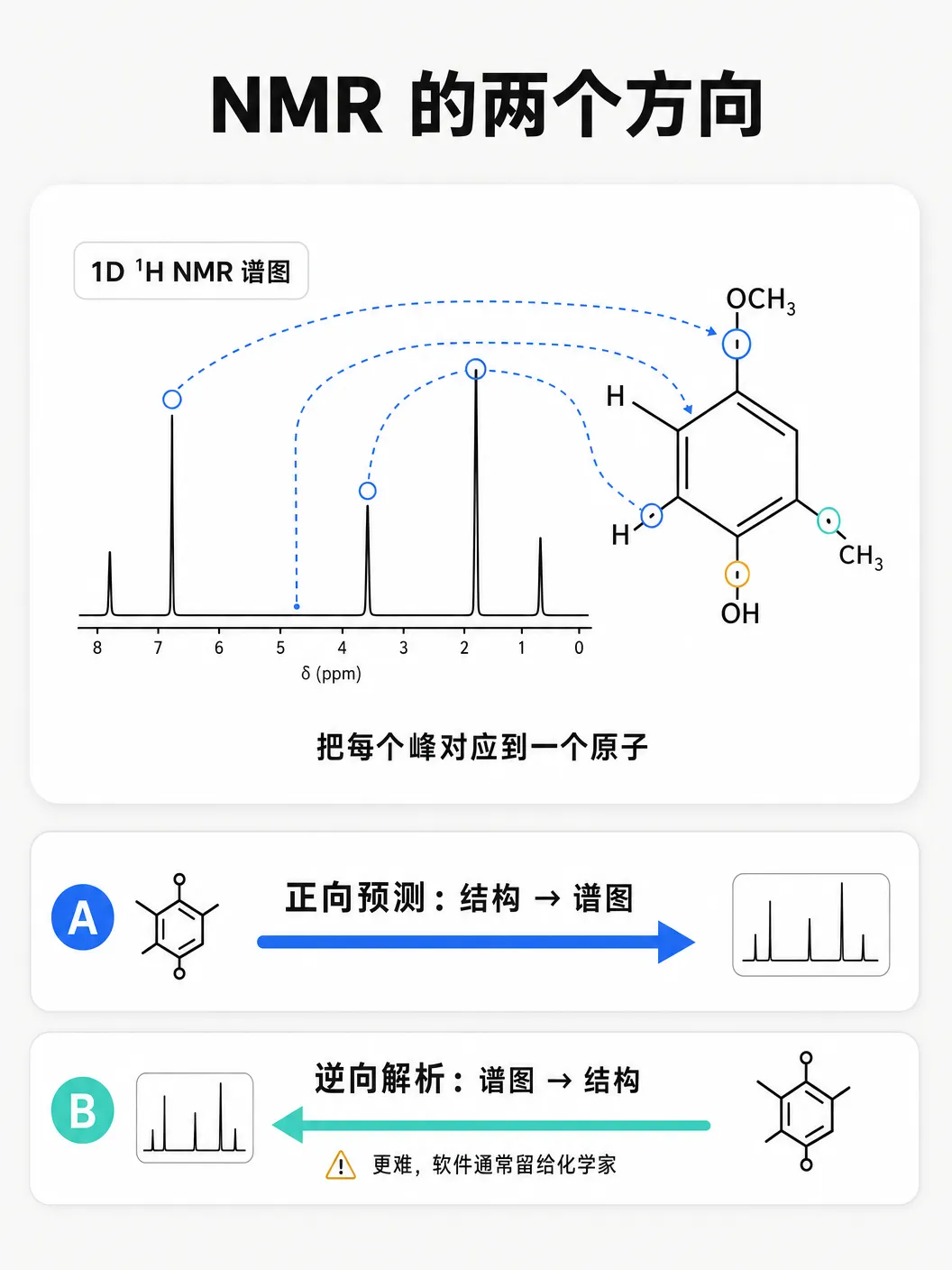
测试沿两个方向展开。正向预测(structure → NMR)是日常活:画出预期结构,预测谱图,再和实测对照——ChemDraw 和 MestReNova 就干这个。
逆向预测(NMR → structure)更难:给一张谱图,反推结构,需要专家级推理判断哪些片段存在、怎么连接。ChemDraw 完全没有逆向能力,MestReNova 能把峰指派到已知结构,但不会从峰列表生成候选结构。
评分协议决定可信度:锁样本、匈牙利匹配、跑三次
结论可不可信,全压在评分协议上。
为避免选择偏差,化合物在生成预测前就被锁定。来源是 ChemRxiv 上的合成化学预印本:化学家逐篇阅读,从补充材料里取每篇第一个完整表征、数据自洽的新化合物。
排除以旋转异构体混合物形式报告的样本,手动转录峰列表,提取 SMILES、¹H 和 ¹³C 峰列表、NMR 溶剂。正向集 20 个、逆向集 15 个,各自独立抽取。

正向集是四类骨架各五个:P1 氯哒嗪(DMSO-d₆ 里慢交换的 NH)、P2 Boc-N-芳基马来酰亚胺与 N-Boc 烯酰胺、P3 螺酮(带苯甲酰甲基或乙酰基侧链)、P4 α-硅基甲磺酰胺(屏蔽的硅-α 碳)。每类专门考一种 NMR 难点。
每个工具拿到一个 SMILES,要在原文献的溶剂里预测 ¹H 和 ¹³C 位移,带多重性和耦合常数。三个 Claude 模型每个化合物各跑 3 次以表征输出波动;ChemDraw 和 MestReNova 是确定性的,每个化合物跑 1 次。
预测峰与实验原子用最小 |Δδ| 做匈牙利一对一指派。宽于 0.3 ppm 的 ¹H 多重峰从位移误差里剔除,保留用于多重性评分。最终对固定分母——401 个 ¹H 原子、225 个 ¹³C 原子——报告 MAE、RMSE、中位 |Δδ|。
核心指标是落在容差窗内的原子比例:¹H ±0.20 ppm、¹³C ±1.0 ppm。
跑三次是为了对付波动。逐化合物的胜者在 20 个样本间频繁易主,单次评测会严重误报胜率;而 Opus 4.7 的 run-to-run 波动小于它和下一名的差距。
正向预测:位移、多重性、耦合常数三项全拿下
¹H 位移,Opus 4.7 的 MAE 是 0.079 ppm,所有受测工具里最低,远在化学家 ±0.20 ppm 的容差窗内。
¹³C 上,Opus 4.7 的 1.37 ppm 与 MestReNova 的 1.48 ppm 基本打平,明显好过 ChemDraw 的 2.107 ppm。
逐化合物胜率,Opus 4.7 在 ¹H 上赢 11.7/20、¹³C 上赢 8.0/20,都是第一。
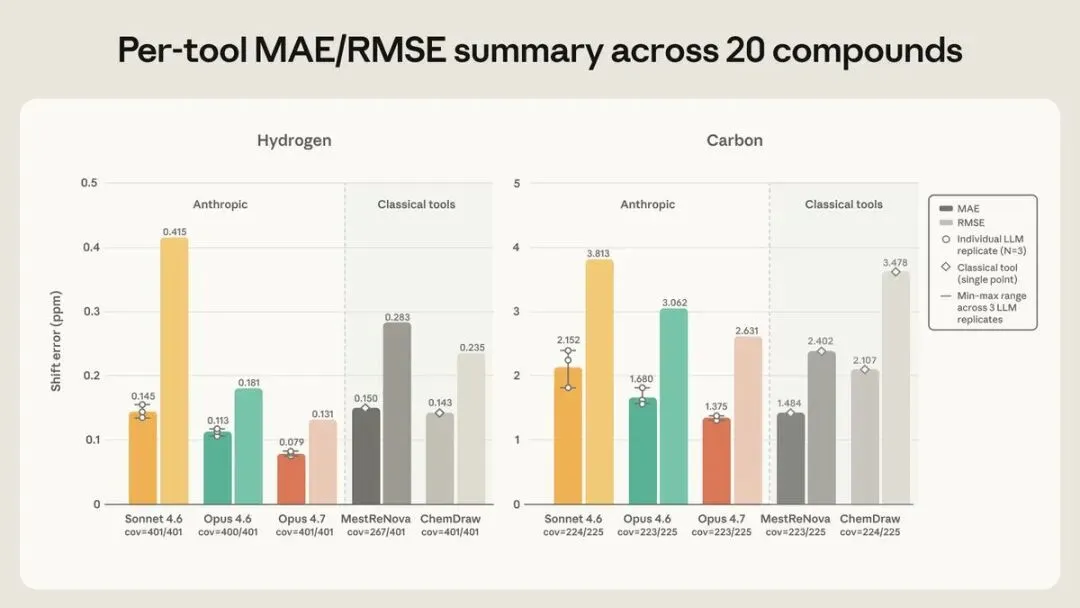
Anthropic 官方 Figure 2:左 ¹H、右 ¹³C 的位移误差(MAE 实心、RMSE 斜纹),Claude 三模型取 3 次均值带波动范围,经典工具为单点。
多重性和耦合常数上,差距更明显。Opus 4.7 的多重性 exact 命中率 85%(343/401),是所有工具最高;MestReNova 54%、ChemDraw 41%。
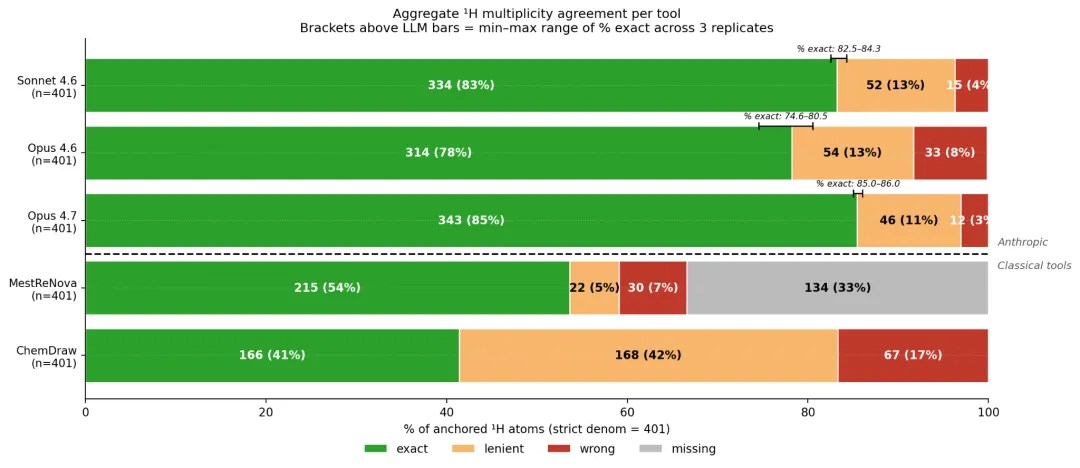
Anthropic 官方 Figure 7:绿 = exact 完全命中、黄 = lenient 实验只报 m、红 = wrong 判错、灰 = missing 没预测到该原子;分母固定 401。
J 耦合上,三个 Claude 模型的平均误差都在 0.5 Hz 上下,落在 ±0.5 Hz 内的比例达 80–84%;经典工具平均误差 1.9–2.0 Hz,±0.5 Hz 内只有 26–35%。
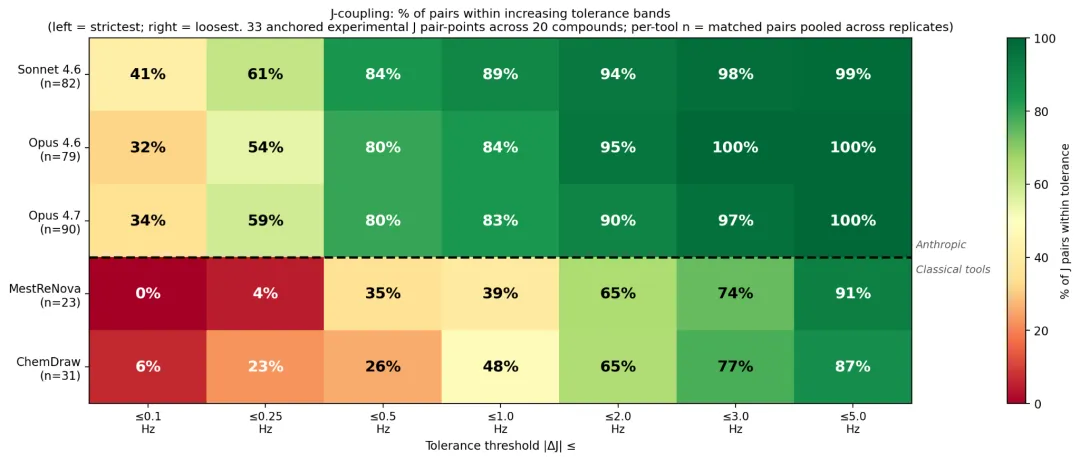
Anthropic 官方 Figure 8:横轴从左(最严 ≤0.1 Hz)到右(最松 ≤5 Hz),格子是落在该容差内的 J 配对比例,绿高红低。Claude 三行明显比经典工具两行绿。
ChemDraw 的 J 值偏差很大程度来自模板默认值:12.4 Hz geminal 耦合在 31 个 J 里出现 5 次,芳香邻位耦合一律发到 7.0–7.1 Hz。
慢交换的 NH 质子实验上落在 6.8–7.9 ppm 的窄窗。Opus 4.7 略偏上场,Opus 4.6 散开几个 ppm,Sonnet 4.6 直接错放到 10–13 ppm。
还有一个所有工具共有的系统性偏差:羰基碳 δ(C=O) 都被预测得偏低——拿”两个工具是否一致”做正确性检查也照样漏。
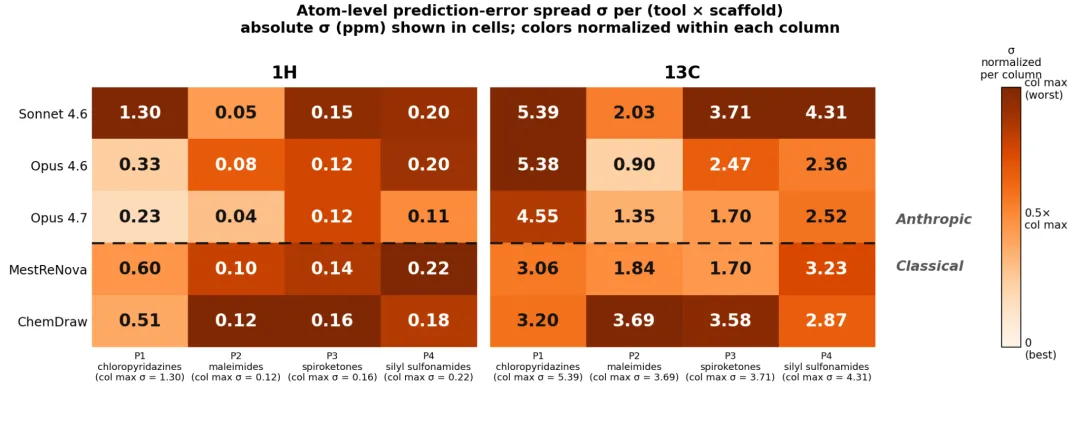
Anthropic 官方 Figure 6:每格是该工具在该骨架内残差的标准差 σ(越小越一致),颜色按列内归一化,越深越差。Opus 4.7 在多数骨架上 σ 最低。
MestReNova 的 ¹H MAE 有个口径差异:它只覆盖了 267/401 个原子,跳过了 >0.3 ppm 的实验多重峰,分数和全覆盖的工具不能直接比。ChemDraw 反而强在覆盖最广。
逆向解析:从谱图反推结构,软件做不了的事
逆向集 Opus 4.7 拿到 15 个 NMR/HRMS 解析问题,每个跑 3 次,要求返回最多 3 个排序的 SMILES 候选。立体化学按设计排除——一维 NMR 定不了绝对构型。
15 个问题分两种 prompt 条件,对应化学家真实面对的两种情形。8 个较简单的目标(Q1–Q5、Q9、Q10、Q14)只给高分辨质谱(HRMS)和一维 NMR,相当于确认一个未指定反应的产物。
7 个结构更密集的目标(Q6–Q8、Q11–Q13、Q15)额外给起始原料 SMILES 作锚点,但不给任何其他反应上下文(无试剂、条件、机理、产物类别),相当于确认一个已知输入的反应产物。
结果:8 个简单目标仅凭谱图和质谱,每次都恢复出已发表结构。7 个密集目标在起始原料 SMILES 下,Q6、Q8、Q12、Q15 三次全对,Q7、Q11、Q13 三次对两次。
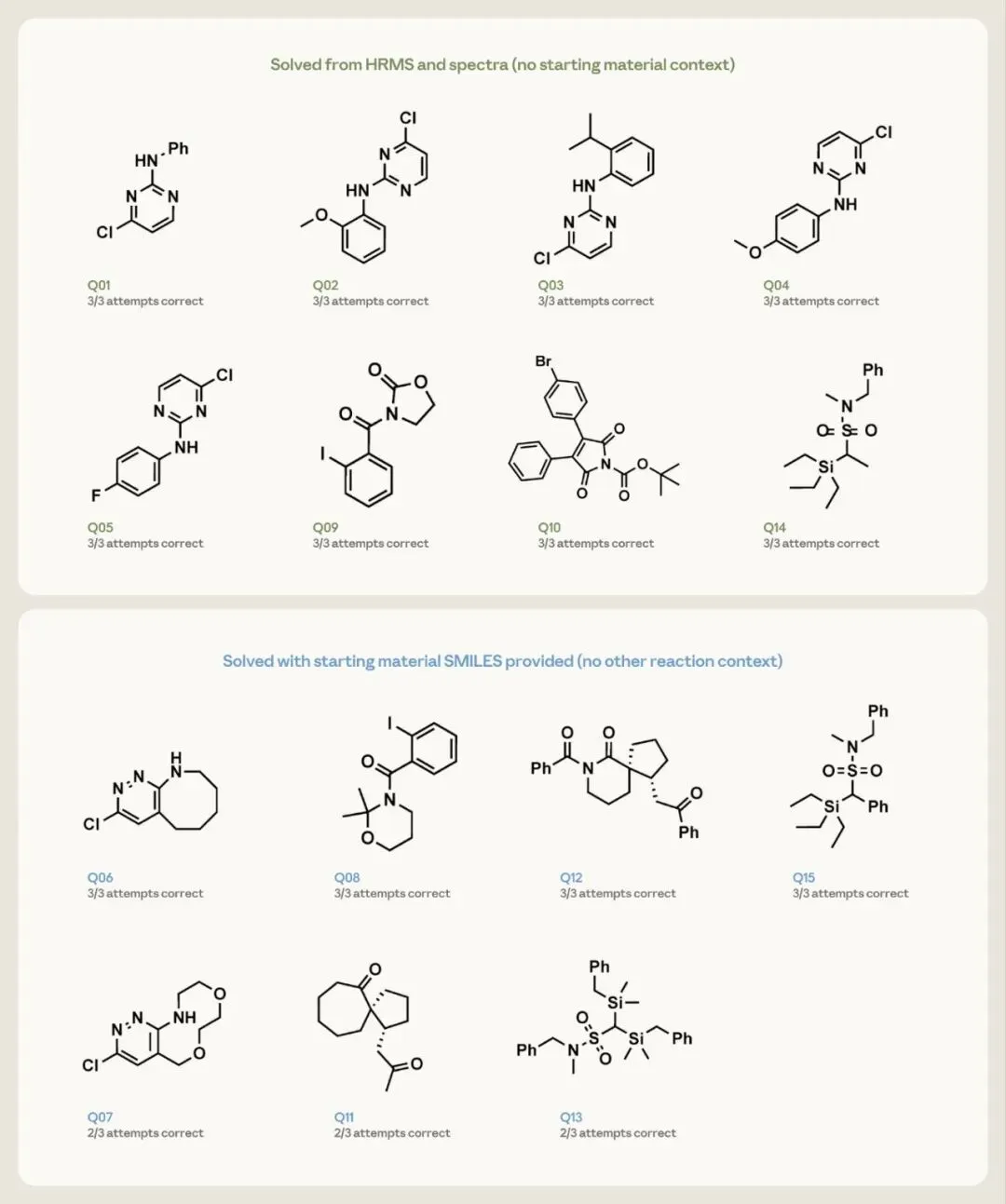
Anthropic 官方 Figure 9:每个分子标注 3 次尝试的命中数。绿框 = 仅凭 HRMS + 一维谱解出(无起始原料上下文),蓝框 = 额外给起始原料 SMILES。
专用结构解析软件存在几十年,通常需要 2D NMR(COSY、HSQC、HMBC 等二维谱),或先假设候选结构再与谱图对照。Opus 4.7 用的只是化学家随手粘进对话框的一维峰列表和质谱。
自动结构解析从依赖 2D 实验的区间,推进到了原本只能靠手工判断的骨架密度。
AI for Science 关键信号:多模态加显式推理,绕开数据壁垒
过去 AI 在化学上难突破,卡点在数据:训练数据稀缺,缺阴性结果、格式不统一、锁在订阅期刊付费墙后。逆合成分析:能用的工具存在多年,普通学术实验室至今不用。
前沿模型是多模态、能显式推理的,可以直接从期刊插图或手绘草图读出结构,不再依赖预先整理好的分子数据库。通用模型不靠领域专用数据库,照样能在专业仪器分析上打平专用软件,还能干软件干不了的逆向题。
变化落在能向一个通用推理模型提的问题上。确认一个已知反应的产物、排除某个区域异构体、对峰指派做 sanity-check、判断哪些化合物值得上 2D 实验——以前要么需要专用软件,要么需要专家逐个判断。现在一个模型用纯文本就能逐项处理。
入口在变,壁垒在评测,化学只是开始
专业软件不会消失,但默认入口会变。ChemDraw、MestReNova 积累了几十年可靠性,短期替不掉。但当一维谱加质谱粘进对话框就能拿到位移、多重性、J 值和一个结构候选,新一代化学家的第一反应会先打开聊天框。

壁垒会从模型本身转移到评测协议。报告最硬的部分是方法学:怎么锁样本、怎么做匈牙利匹配、为什么跑三次、用哪个固定分母。模型能力是公共供给,能拉开差距的是有没有一套严谨、可复现、骗不过去的领域评测。
化学只是开始。多模态读图、显式推理、不依赖封闭数据库——同一套逻辑可以平移到材料、生物、药物。一旦在一个学科证明跑得通,复制成本很低。
20 个化合物之后,真正的考验才开始
样本小:正向 20 个、逆向 15 个,每个骨架只贡献单一类失败模式,数值排名只能指示方向,不是精确结果。慢交换 NH 杂芳烃只通过氯哒嗪采样,相关的羟基吡啶、氨基噻唑等 DMSO-d₆ 里 NH 活泼的体系都没测。
2D 实验(COSY、HSQC、HMBC)和立体化学按设计排除。溶剂只覆盖 DMSO-d₆、CDCl₃、D₂O,甲醇-d₄、苯-d₆、丙酮-d₆ 未评估。
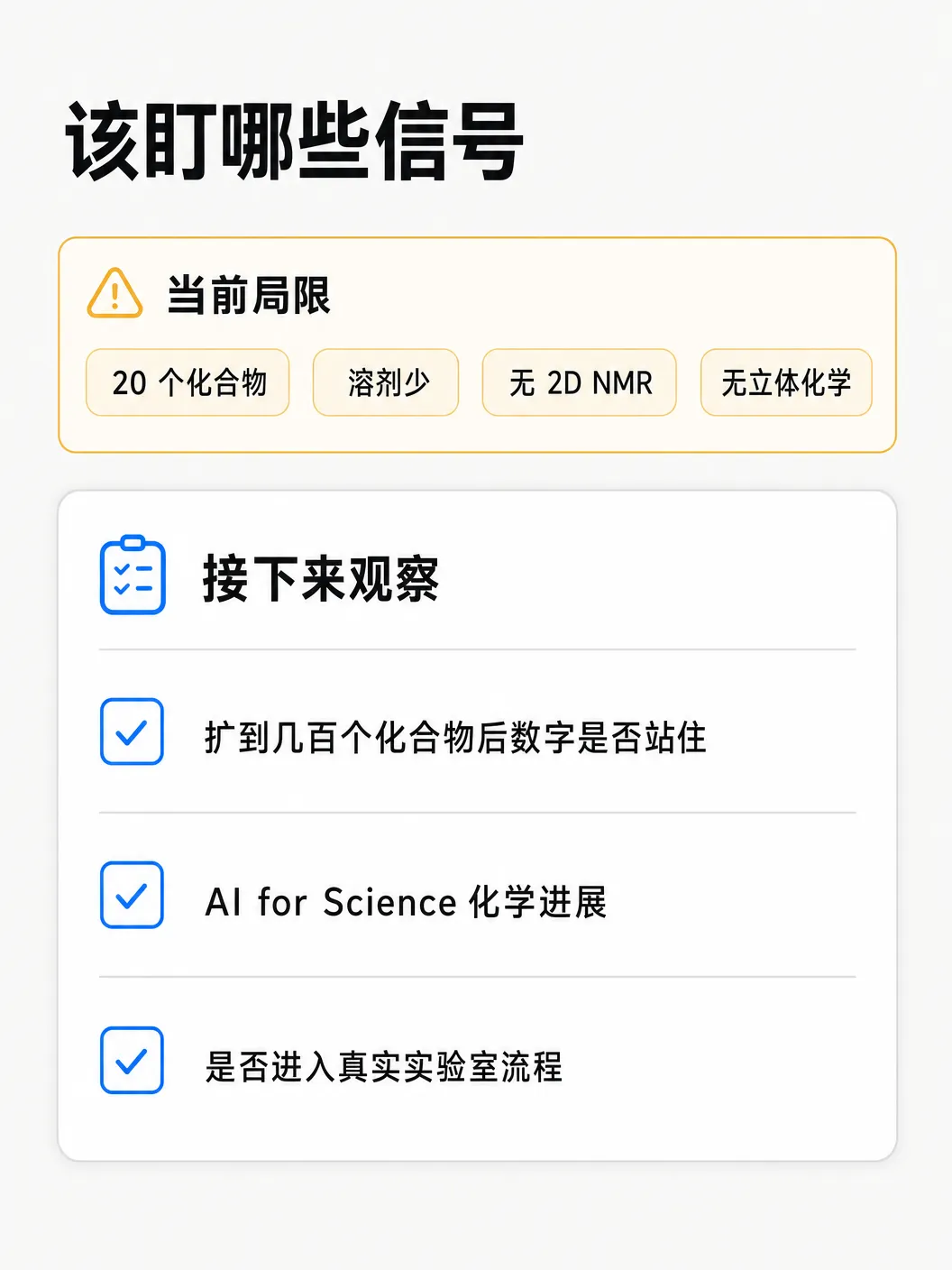
接下来值得盯三件事:扩到几百个化合物、横跨二三十类骨架后准确率会不会塌;逆向解析能不能在没有起始原料锚点时啃下更密的结构;能力会不会真的进到实验室日常流程,被当成每天打开的工具。
20 个化合物的盲测规模不大,但已经划出一条线:通用大模型在专业仪器分析上打平了几十年的窄域软件,还接下了软件做不了的逆向题。小样本上的成绩有了,下一道坎是它能不能扛住更大规模,从一次演示变成化学家每天打开的对话框。
参考信息:
https://www.anthropic.com/research/making-claude-a-chemist
https://www-cdn.anthropic.com/07441e654ad3dfeb0cd090e9361511562825d012.pdf