 夜雨聆风
夜雨聆风软件介绍
LAMMPS是一款经典的分子动力学代码,专注于材料建模。它是大规模原子/分子并行模拟器(Large-scale Atomic/Molecular Massively Parallel Simulator)的缩写。
LAMMPS在固态材料(金属、半导体)和软物质(生物分子、聚合物)以及粗粒化或介观系统中具有应用潜力。它可用于模拟原子,或者更一般地,作为原子、介观或连续尺度上的并行粒子模拟器。
注意事项
⏩:测试环境win11/24H264位操作系统
⏩: 部分软件为了避免误杀小白 可直接 安装火绒杀毒 接管系统自带的Defender杀毒软件
⏩: 电脑 用户名 和 软件安装路径最好是英文
软件测试
1、右键解压
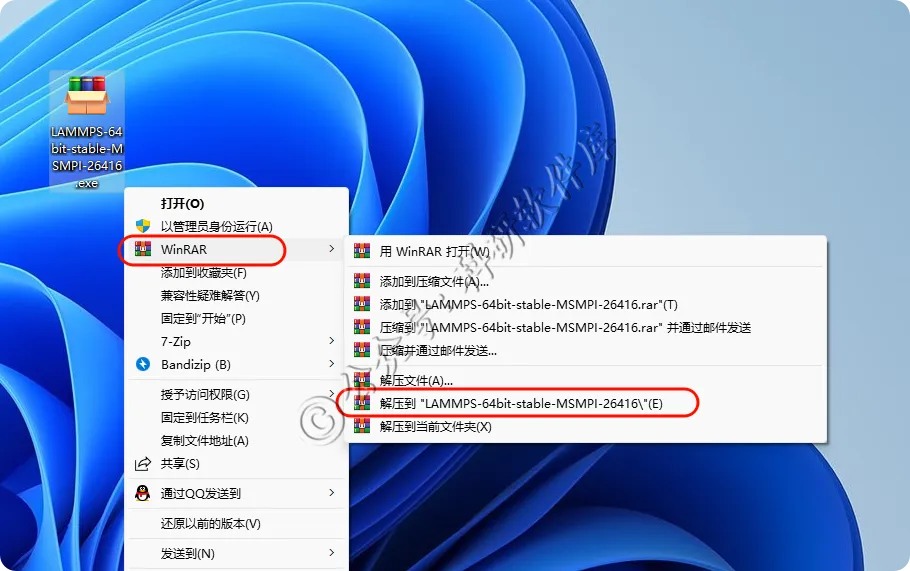
2、进入解压后的文件夹

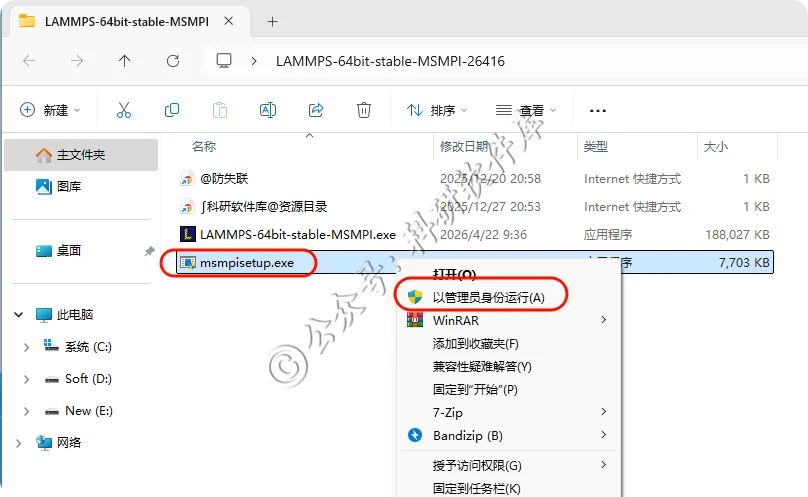
3、右键管理运行msmpisetup.exe安装程序
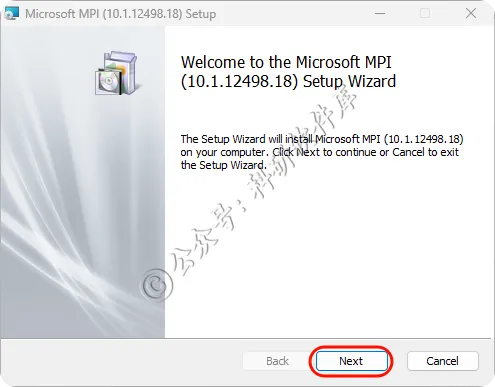
4、点击Next
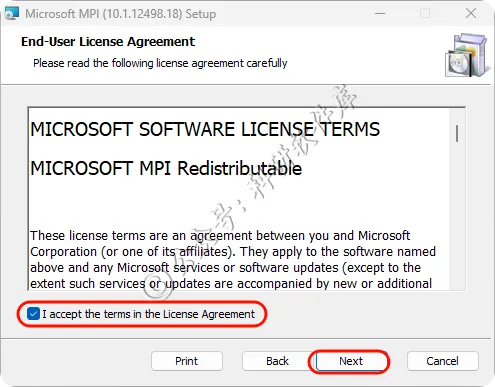
5、勾选I accept...点击Next
6、点击Next
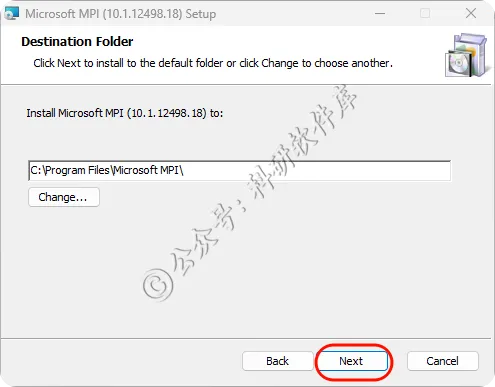
7、点击Install
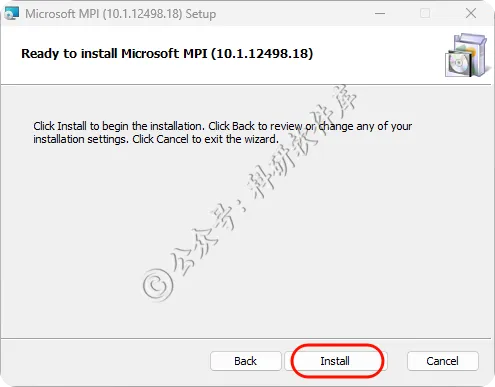
8、点击Finish
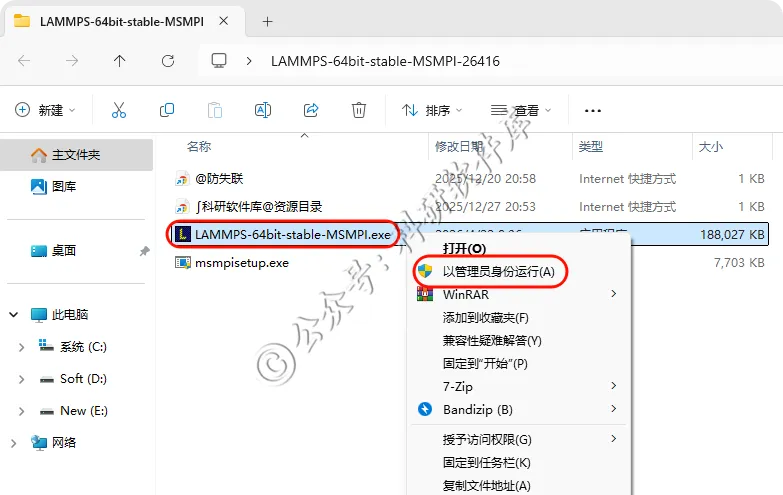
9、右键管理运行LAMMPS...
10、点击I Agree
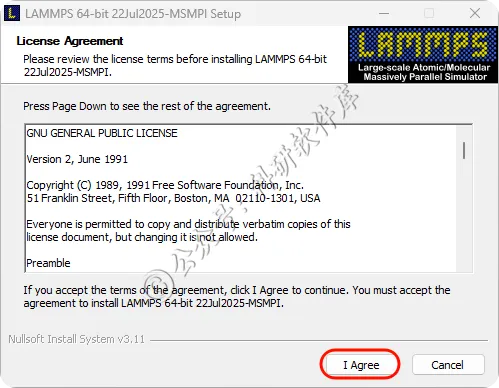
11、点击Install
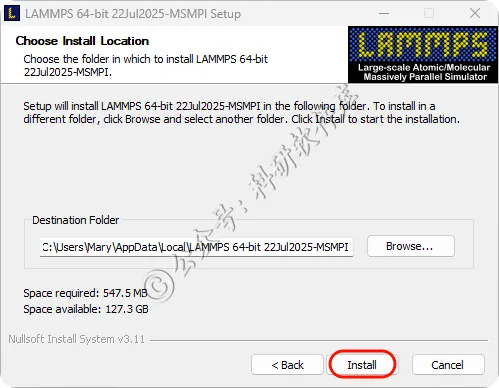
12、等待程序安装
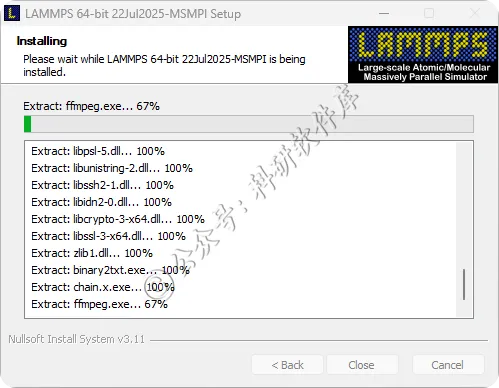
13、点击Close
14、右键管理运行命令提示符
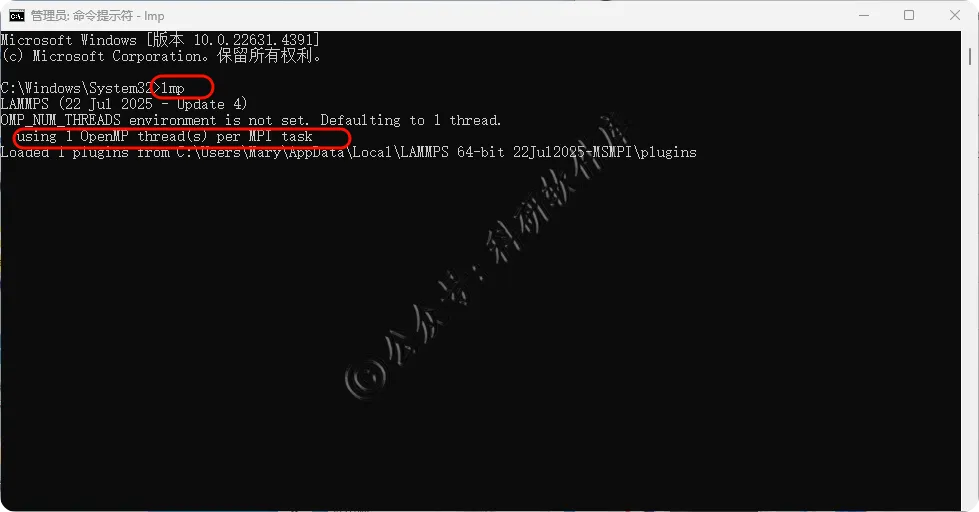
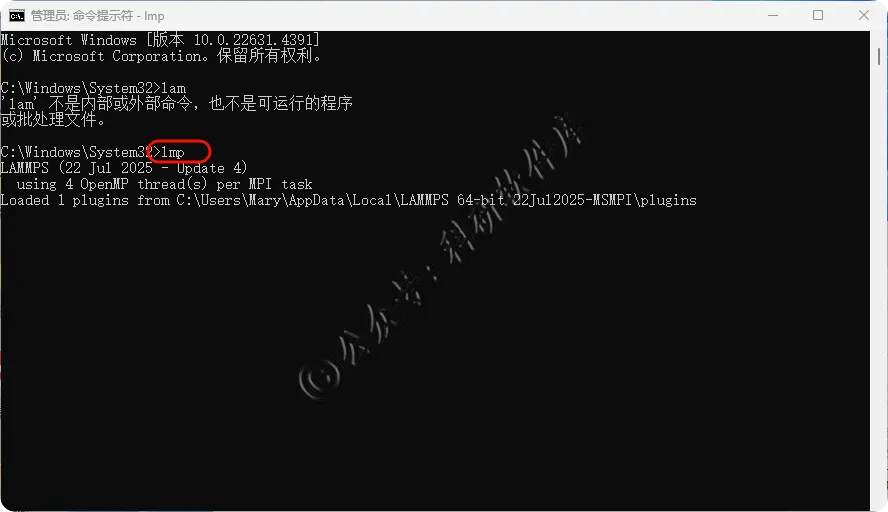
15、输入lmp 出现版本号 软件安装成功 如若提示using 1 OpenMP thread(s) per MPI task可以手动设置
16、右键此电脑—属性
17、点击高级系统设置
18、点击环境变量
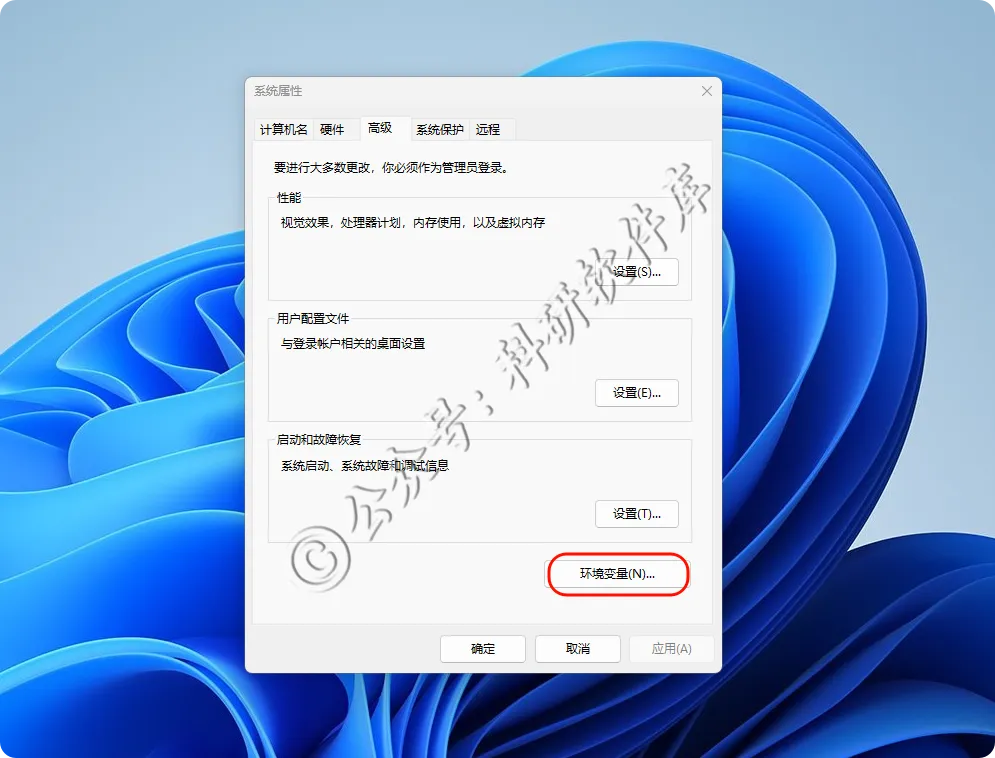
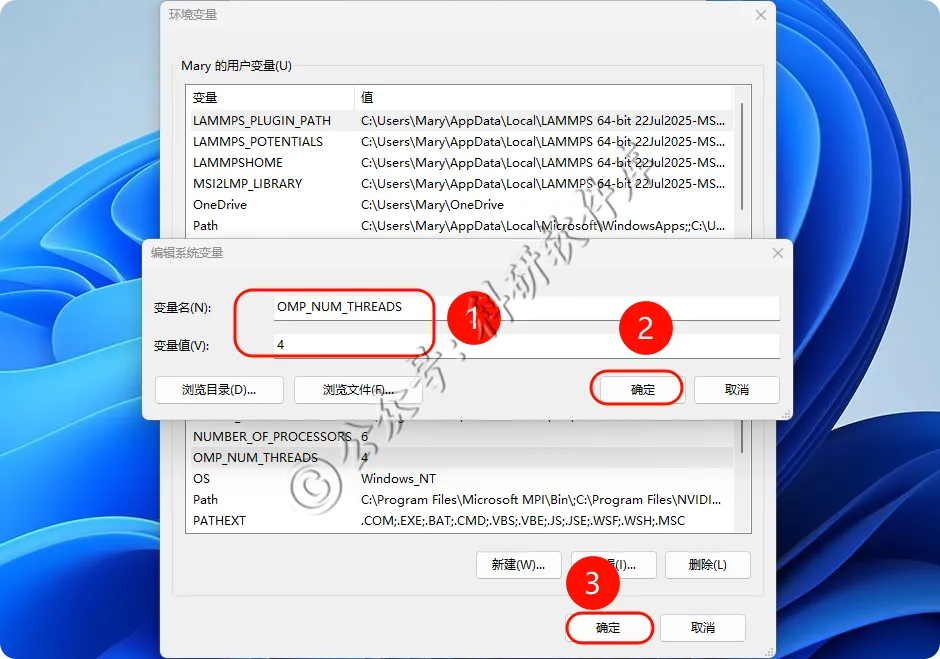
19、变量名OMP_NUM_THREADS 变量值为核心数不能超过自己电脑核心 小编设置的为4 点击确定
20、再次输入lmp 提示using 4 OpenMP thread(s) per MPI task
资源获取
🔑 公众号回复「L03」获取下载。
