 夜雨聆风
夜雨聆风
核心计算逻辑
在体系原子数、k点、截断能参数固定的前提下,AIMD每一个离子步时长基本趋于稳定,因此采用单步平均耗时 × 总模拟步数的精准预估方案,公式如下
AIMD总耗时(s) = 单离子步平均耗时(s/step) × 总模拟步数(step)AIMD总耗时(h) = 总耗时(s) ÷ 3600
其中总模拟步数由INCAR中的NSW参数直接定义,是耗时预估的核心基数
前期准备:完整输入文件与脚本配置
耗时预估前,需提前配置好全套AIMD计算输入文件,保证参数与正式计算完全一致(参数不一致会导致单步耗时偏差,预估失效)
所需文件包含:INCAR、KPOINTS、POSCAR、POTCAR、超算提交脚本sub_vasp.sh
核心输入文件参数示例
1. INCAR文件
固定AIMD模拟参数,此处为常规常温NVT系综分子动力学参数,可直接复用:

本次示例核心参数:总步数NSW=10000步,后续预估以此为基准
2. 其余基础文件
POSCAR:待模拟体系的初始结构文件(原子数量、晶格参数固定)
POTCAR:对应体系元素的赝势文件,与POSCAR元素匹配
KPOINTS:k点网格文件,常规分子动力学采用(1×1×1)即可,参数统一固定
3. 超算提交脚本 sub_vasp.sh 示例
脚本资源申请参数(CPU、核数、内存)必须与正式计算保持一致,资源差异会直接改变单步耗时
#!/bin/bash#SBATCH -p normal#SBATCH -J AIMD-time-test#SBATCH --nodes=1#SBATCH --ntasks=1#SBATCH --gres=gpu:1module load vasp/6.2.0mpirun vasp_std
分步耗时预估实操完整流程
步骤1:提交测试任务
将全套输入文件与提交脚本上传至超算工作目录,在终端执行提交命令,提交测试任务
sbatch sub_vasp.sh
此处重点:测试任务无需跑完所有步数,仅需要程序正常迭代跑出30~50个离子步即可,步数过少会导致平均误差大,步数过多浪费算力
等待超算运行5~20分钟,确保OUTCAR文件中生成完整的离子步迭代记录
步骤2:抓取离子步运行耗时数据
任务运行稳定、跑出足够离子步后,取消任务,使用命令抓取OUTCAR中每一步LOOP+迭代的计算耗时
grep LOOP+ OUTCAR
命令输出结果示例
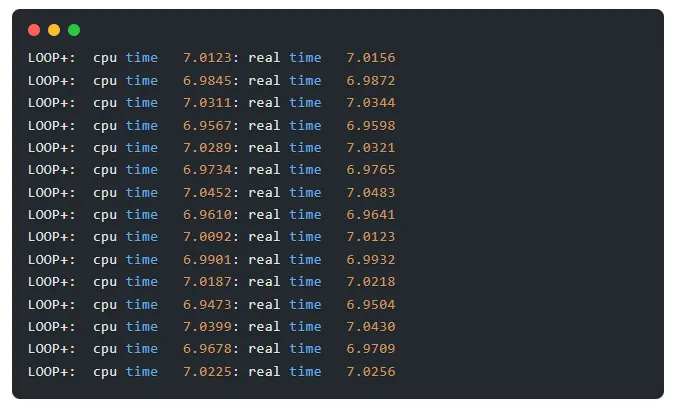
输出中 real time 后的数值为该离子步的实际运行耗时(单位:秒),是我们的统计依据
步骤3:计算单步平均耗时
直接统计全部 15 组耗时数据求取平均值
耗时数据:7.0156、6.9872、7.0344、6.9598、7.0321、6.9765、7.0483、6.9641、7.0123、6.9932、7.0218、6.9504、7.0430、6.9709、7.0256
总耗时合计 = 105.0352s
单步平均耗时 = 105.0352 ÷ 15 ≈ 7.00 s/step
步骤4:计算AIMD模拟总耗时
结合本次INCAR参数:总步数NSW=10000步,代入核心公式计算
总耗时(秒)= 7.00 s/step × 10000 step = 70000 s换算为小时:70000 ÷ 3600 ≈ 19.4 小时
实操关键注意事项(避坑重点)
- •
参数一致性原则:测试任务与正式任务的INCAR参数、k点、截断能、原子体系、超算核数/内存必须完全一致,任意参数改动都会改变单步耗时,导致预估失效
- •
剔除异常步数:OUTCAR中前1~2步为程序初始化迭代,耗时不稳定,必须剔除后再计算平均值,提升预估精度
- •
取样步数要求:建议取样30步以上有效数据取平均,体系越大、原子数越多,需要的取样步数越多,避免偶然误差
五、批量统计快捷命令(进阶实操)
若步数较多,手动统计繁琐,可直接使用以下命令快速计算平均单步耗时,无需手动计算
grep LOOP+ OUTCAR | tail -n +3 | awk'{gsub(/:/,"",$7); sum+=$7; count++} END {print "平均单步耗时:", sum/count, "s"}'
命令功能:自动剔除前2步数据,统计剩余所有LOOP+步数的平均耗时,直接输出结果
温馨提示:如需取用代码,建议访问官网 www.yuansuan.top,查阅配套帮助文档就能轻松复制代码。
有需要算力租用、服务器定制和集群搭建的老师们,欢迎通过以下方式联系我们!

扫描二维码添加好友
联系邮箱:info@wfyskj.com.cn
电话:17616746026
