 夜雨聆风
夜雨聆风Listgarten, J., & Jiang, H. (2026). How artificial intelligence is reengineering protein engineering. Science, 392(6794), 159-166.
https://www.science.org/doi/10.1126/science.aec8444
摘要(Abstract)
在过去的几十年里,蛋白质工程已经发展成为一个独立的领域,受计算建模和高通量湿实验室实验的驱动,在治疗、诊断、农业和制造业中具有广泛的应用。近年来,人工智能(AI)通过能够在高维序列空间中更高效地搜索具有所需特性的蛋白质,进一步推动了蛋白质工程的发展。基于AI的显著进展包括序列、主链结构和原子的生成建模;针对设计特定特性蛋白质定制这些模型的通用版本;提取蛋白质表征和对候选蛋白质序列进行打分的建模;以及开发包括感知合成方法在内的文库设计技术。在本文中,我们讨论了这些进展,强调通过对现代AI方法的统计学解释提供一个统一的视角。
科学问题与研究背景
蛋白质是生命的基石,无论是催化能量代谢还是提供细胞结构,几乎所有的生物学过程都离不开它的参与。人类很早就开始尝试“改造”蛋白质以开发新药、改良农作物或制造新材料。然而,大自然用了数十亿年才进化出当下的蛋白质网络,人类却希望在几年甚至几天的极短时间内“按需定制”蛋白质。
在人工智能全面介入之前,蛋白质工程有两大主流路径:定向进化(DE)与计算蛋白质设计(CPD)。定向进化通过多轮突变和人工筛选,在湿实验中逐步逼近目标,但它极其依赖一个已经具备初步功能的“初始蛋白质”,且实验成本高昂、通量有限;计算蛋白质设计(CPD)则完全基于物理能量函数在计算机内探索空间,虽然不依赖完整的初始序列,但其粗糙的能量函数往往无法捕捉酶催化所需的复杂动态或量子力学效应。
这两大传统方法都撞上了同一堵叹息之墙——极度庞大的搜索空间。一条仅由100个氨基酸组成的小型蛋白质,其可能的序列组合就高达 20的100次方(约等于 10的130次方),这一数字甚至远远超过了可观测宇宙中的原子总数(约 10的80次方)。更致命的是,在这片浩瀚的序列海洋中,仅有极小一部分变体能够成功折叠并表达。面对如此庞大的维度灾难,传统的随机突变或单一物理引擎打分显然力不从心。如何更高效地进行大跨度的“智能跳跃”,并快速、低成本地预测候选序列的特性?这正是现代AI技术试图攻克的终极科学难题。
核心逻辑与深度解析
为了解决传统方法中犹如“盲人摸象”般的低效搜索,研究人员开始将目光转向机器学习,特别是概率与生成模型。最直观的改造是:利用计算机内的预测模型对序列的适应度(如催化效率)进行打分。但这依然不够,如何聪明地提出突变序列?答案在于生成模型。
早期的计算机内进化算法只是盲目突变,而现代AI引入了变分自编码器(VAE)等生成模型来提议突变。随着优化迭代,生成模型能够捕捉到高分序列的分布规律,甚至学会“协同突变”(即考虑氨基酸之间的上位效应),从而逐步“嗅探”出适应度景观的几何结构。这就将传统的“搜索特定序列”问题,降维打击并转换为“寻找目标概率分布的参数”问题。
然而,单纯依赖预测模型容易在未见过的序列空间中失准。为了解决这一偏差,研究人员引入了“自适应采样条件化”(CbAS)策略。这一策略本质上是在执行经典的贝叶斯定理:结合编码了泛蛋白质先验知识的无条件生成模型,与通过实验数据训练的属性预测模型,从而得到满足特定功能要求的条件概率分布。在这个框架下,设计具有特定属性的蛋白质,就等价于从这个复杂的条件分布中进行精准采样。
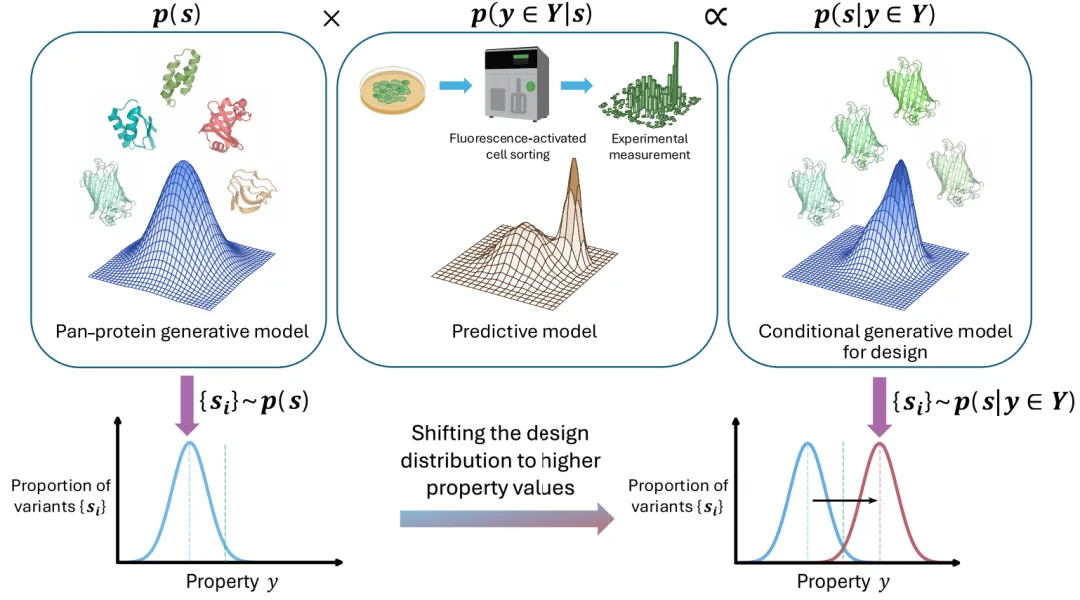
[图1:基于统计学视角的AI蛋白质工程图解]
蛋白质序列设计本质上等同于从目标条件分布 p(s|y属于Y) 中采样序列 s。图中展示了通过结合已有知识进行设计的策略:将编码为预训练生成模型 p(s) 的背景知识,与通过实验数据构建的属性预测模型 p(y属于Y|s) 相结合。这一结合过程遵循贝叶斯定理,即条件分布正比于两者的乘积。通过流式细胞术等实验测量获取数据,模型能够将设计分布向高于阈值(绿色虚线)的更高属性值区域推移,从而大幅提升采样出高活性或高稳定性候选突变体的概率。
明确了统计学目标后,AI模型的设计架构演化出了三种截然不同的获取条件生成模型的路径。第一种是“硬编码”式,在训练之初就把酶学委员会编号、二级结构等条件变量融入模型,缺点是灵活性差,一旦需要新条件就得重训庞大的模型。第二种就是前文提到的贝叶斯规则结合法,它赋予了模型“即插即用”的优势,但在数学上需要处理难以计算的分母项(对所有可能序列求和)。为了绕开这个计算黑洞,第三种“即时引导(On-the-fly)”策略应运而生。扩散模型和流匹配模型便是其中的佼佼者。它们不直接估计概率密度,而是去估计对数概率密度的梯度。在逐步去噪的采样过程中,模型可以直接用属性预测模型的梯度来“引导”无条件模型的生成方向,完美避开了复杂的分母计算。
在这些底层逻辑的支撑下,专用的模型架构如雨后春笋般涌现,重构了从主链到序列的整个设计流:
从主链生成到序列反推:传统的从头设计往往是先生成3D主链,再推导序列。Chroma和RFdiffusion等在PDB全库上训练的扩散模型,成功实现了通过即时引导生成极其复杂且符合物理规律的主链结构。有了主链后,逆向折叠模型(如ProteinMPNN)接管了工作。实验数据显示,在给定主链推导序列的任务中,ProteinMPNN的序列恢复率达到了 52.4%,全面碾压了传统Rosetta的 32.9%,证明了图神经网络在捕捉三维几何空间特征上的巨大优势。
全原子与多模态联合生成:分步设计依然存在误差累积的问题,最新的趋势是直接进行序列和结构的全原子联合建模。像ESM3这样的多模态大语言模型,甚至整合了序列、离散化结构以及自然语言功能注释三大模态,让模型能够跨维度共享信息。
尽管AI取得了惊人成就,但实验验证环节暴露出新的两极分化。在蛋白质结合剂的设计上,AI带来了颠覆性的突破:前AI时代,计算设计的结合剂文库命中率通常低于 0.05%,而现在借助生成模型,命中率提高了数个数量级,甚至可以通过微孔板进行常规表征,彻底告别了劳动密集型的高通量筛选。然而,针对酶工程这种需要精确到原子级别和量子力学计算的领域,AI目前仍步履维艰,大多只能通过提取已知酶的活性位点进行条件生成,难以直接创造出催化全新反应的酶。
总结与展望
本文全景式地复盘了AI如何利用生成模型与贝叶斯条件化策略,将庞杂无序的蛋白质搜索难题转化为高度结构化的概率采样过程。其最大的突破在于,不仅提供了生成新序列的能力,更构建了一套以即时梯度引导和多模态对齐为核心的“定向降维”工具箱,极大地提升了功能蛋白质的设计命中率。
放眼未来,当前的AI设计依然高度依赖AlphaFold等结构预测工具作为事后过滤的评判标准,但这存在陷入“天然蛋白质流形偏见”的风险。蛋白质工程亟需建立更稳健的纯计算机内(in silico)基准测试环境,并将放之四海而皆准的物理生物学模型与强于局部插值的AI模型深度融合。只有当计算设计的评估不再完全受限于高昂的湿实验验证成本,蛋白质工程才能真正迈入“可编程生命”的新纪元。
作者介绍Jennifer Listgarten 现为加州大学伯克利分校电气工程与计算机科学系(EECS)、计算生物学中心教授,同时任职于加州大学伯克利分校与旧金山分校(UCB-UCSF)联合生物工程研究生项目。Hanlun Jiang 为其团队核心研究人员,双方致力于将统计机器学习理论应用于计算生物学与蛋白质工程前沿问题。
