 夜雨聆风
夜雨聆风





重磅|Nature颠覆生物催化!AI工具CATNIP直接连接化学空间与蛋白序列,一键预测酶-底物配对
想做一步生物催化反应,却不知道该用哪个酶、能转化什么底物? 传统方法只能在已知反应附近“小修小改”,99.7%的序列功能未知,海量潜力被深埋。
密歇根大学 + 卡内基梅隆大学团队在Nature发表终极方案: 先通过高通量实验建立215个全新酶-底物配对,再训练AI模型CATNIP,实现底物→推荐酶 / 酶→推荐底物双向精准预测,直接打通化学空间与蛋白序列空间,让生物催化从此“可预测、低风险、高效率”。
一、研究背景:生物催化的世纪痛点
-
制药与合成大势所趋生物催化绿色、高效、选择性强,可缩短合成路线、提升收率。 -
核心瓶颈
-
酶底物范围无法预测 -
海量序列功能未知 -
只能在已知点附近局部探索,无法跨空间跳跃
-
科学问题能否建立通用模型,直接连接“小分子化学空间”与“蛋白序列空间”?
本研究给出答案:能,而且只需两个维度:底物结构 + 酶序列相似度。
二、整体研究内容总览
-
酶家族:α-KG/Fe(II)依赖型非血红素铁酶(NHI),万能C–H官能化 -
酶库:aKGLib1,314个多样性酶,平均同源仅13.7% -
底物库:111个多样分子(药物、天然产物、杂环、脂肪酸) -
发现:215个全新生物催化反应 -
模型:CATNIP(梯度提升树),双向预测 -
底物 → 推荐最可能的酶 -
酶序列 → 推荐最可能的底物 -
验证:训练集/测试集/外部序列三重验证成功
三、逐图深度解析
Figure 1|生物催化发现的现状与本研究范式
核心结论:从“局部摸索”升级为“全域跨空间预测”。
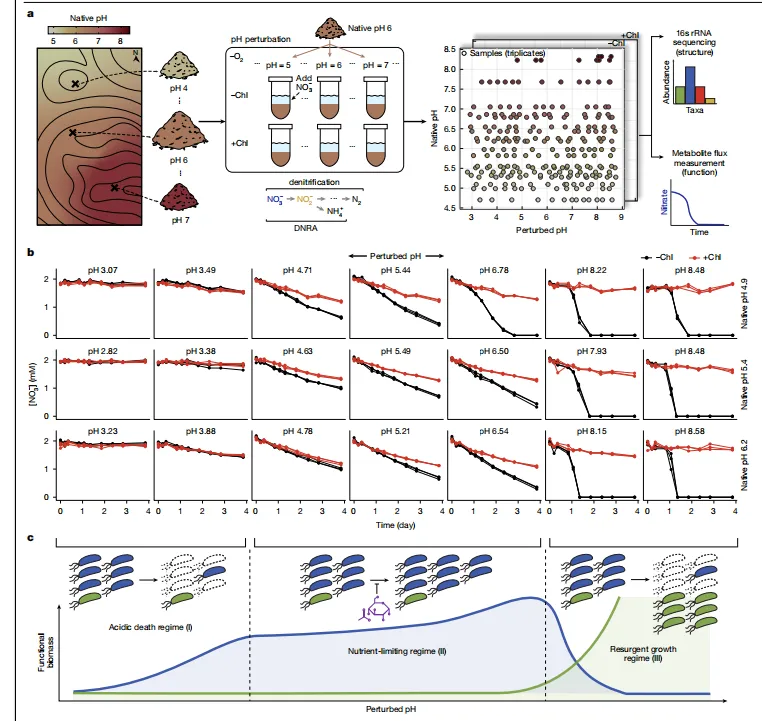
-
传统路线:已知反应 → 局部化学改造 / 局部蛋白突变 -
局限:无法跨越巨大的未探索化学与序列空间 -
本研究路线: -
高通量建立全新酶-底物配对 -
训练模型连接两大空间 -
实现全域、无前提预测 -
目标:彻底降低生物催化在合成路线中的风险与试错成本
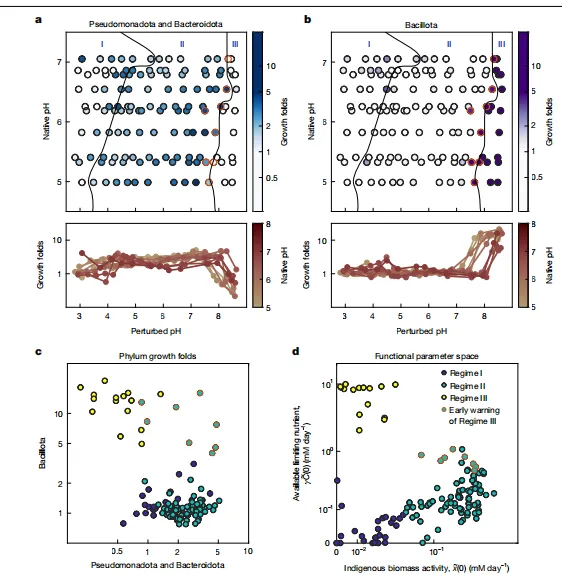
Figure 2|aKGLib1酶库构建:覆盖序列多样性
核心结论:构建最大、最多样的α-KG依赖酶库。
-
基于面部三联体(2His-1羧酸盐)保守位点筛选 -
序列相似网络(SSN)去冗余,得到27,005个序列 -
挑选314个酶: -
102个来自大簇 -
125个未表征 -
87个已知/推测功能 -
平均同源**13.7%**,覆盖巨大序列空间 -
78%可溶表达,30%有已知活性
Figure 3|高通量反应发现:215个全新生物催化反应
核心结论:32%底物被转化,38%酶有活性,以羟化与去饱和为主。
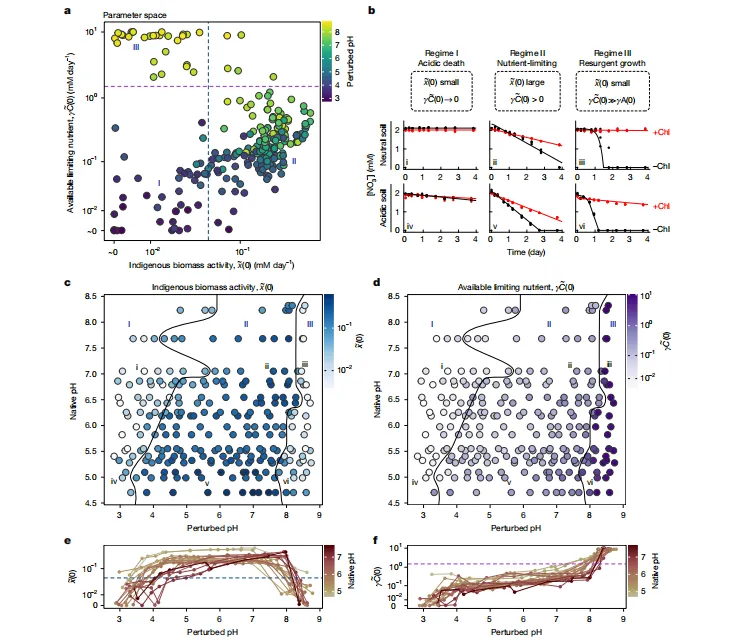
-
96孔板高通量流程:全细胞粗酶 → 加底物 → LC–MS检测 -
检测:羟化、去饱和、重排、氯化 -
结果: -
111种底物 → 35种被转化(32%) -
314种酶 → 119种有活性(38%) -
总计215个新反应 -
反应类型:羟化64%、去饱和18%、混合18% -
底物遍布化学空间,无集中偏向
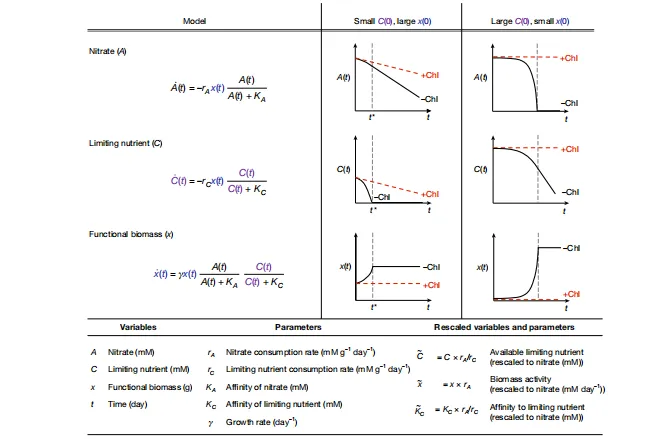
Figure 4|机器学习建模:底物↔酶双向预测
核心结论:用化学空间距离 + 序列相似度,构建高精度推荐系统。
-
数据集BioCatSet1:215新反应 + 139文献反应 -
底物特征:MORFEUS计算21个参数 → PCA降维 -
酶特征:SSN提取序列相似度(AS%) -
双模型: -
底物→酶:找近邻底物 → 取对应酶 → 排序推荐 -
酶→底物:找近邻酶 → 取对应底物 → 排序推荐 -
评价指标:precision@k / recall@k / enrichment@k / nDCG@k -
最优模型:GBM梯度提升树,显著优于基线
Figure 5|CATNIP网页工具实战:训练集/测试集/外部序列全验证
核心结论:输入结构/序列,一键出结果,实验成功率极高。
-
底物→酶预测: -
金雀花碱(16)→ 推荐10个酶 → 7个活性,制备35%收率 -
苦参啶(18)→ 7个活性,制备50%收率 -
甾体烯酮(20)→ 7个活性,首次实现氧化切断 -
酶→底物预测: -
NHI123 → 推荐底物22,7%转化 -
NHI177 → 推荐humulene(12),41%转化 -
外部酶TqaL → 推荐底物23,42%转化 -
结论:跨家族、跨数据集、跨物种通用
四、实验与分析方法流程总结
-
生物信息学构建酶库收集IPR家族 → SSN网络构建 → 去冗余 → 挑选314个序列 -
高通量克隆与表达全基因合成 → pET28b → E. coli 96孔表达 → SDS-PAGE验证 -
高通量生物催化筛选全细胞悬液 → 加底物/α-KG/VC/Fe(II) → 厌氧反应 → LC–MS检测 -
化学空间表征SMILES → MORFEUS计算21个描述符 → PCA -
序列相似度计算SSN比对得分 → 归一化AS% -
机器学习与评估构建近邻推荐 → GBM提升 → 五折划分 → 指标评估 -
网页工具部署CATNIP在线平台:输入SMILES或序列 → 输出排名列表 -
制备规模反应验证1L发酵 → 裂解液反应 → 分离纯化 → 结构鉴定
五、论文核心结论
-
首次实现化学空间 ↔ 蛋白序列空间的全域预测 -
建立314个多样性酶库与215个全新生物催化反应 -
训练CATNIP双模型,支持底物→酶 / 酶→底物双向推荐 -
模型精度高、泛化强,可预测训练集外分子与序列 -
提供免费网页工具,直接用于合成路线设计与筛选 -
范式可扩展到P450、转氨酶、水解酶、氧化还原酶等全家族
六、研究展望与合成革命
-
药物合成路线快速设计直接预测关键中间体的一步酶法合成 -
后期修饰(LSF)药物分子定点C–H羟化/去饱和,快速获得代谢产物 -
天然产物高效合成复杂萜类、生物碱、生物碱的选择性氧化 -
酶工程起点优化从CATNIP推荐出发,大幅减少进化轮次 -
全域生物催化数据库继续扩展酶家族与反应类型,建成生物催化版PubChem
论文信息
题目:Connecting chemical and protein sequence space to predict biocatalytic reactions 期刊:NatureDOI:10.1038/s41586-025-09519-5发表单位:
-
密歇根大学生命科学研究所、化学系 -
卡内基梅隆大学化学工程、化学、机器学习系 -
巴西圣玛丽亚联邦大学 -
斯科特能源创新研究所