 夜雨聆风
夜雨聆风





Cell│北大:通过AI辅助实现肠道微生物胆汁酸代谢酶的鉴定,揭示了肠道菌群对单酸酰化胆汁酸的水解机制
 点击蓝字“代谢组metabolome”,轻松关注不迷路
点击蓝字“代谢组metabolome”,轻松关注不迷路

生科云网址:https://www.bioincloud.tech/
编译:微科盟搬砖人,编辑:微科盟X、江舜尧。
微科盟原创微文,欢迎转发转载。
胆汁酸的修饰对宿主生理与病理过程具有重大意义。鉴定其合成酶对于揭示胆汁酸多样性及开发靶向干预策略至关重要,但这目前仍是一项重大挑战。为解决该难题,本研究开发了人工智能辅助工具胆汁酸酶解析单元,其预测了超过60万个候选胆汁酸代谢酶,并汇总至人类广义微生物胆汁酸代谢酶数据库(https://beaut.bjmu.edu.cn)。本研究鉴定出一系列未表征的胆汁酸酶,包括单酸酰化胆汁酸水解酶和3-乙酰基脱氧胆酸合成酶。值得注意的是,ADS能通过碳-碳键延伸,进而合成出从未被报道过的骨架胆汁酸3-乙酰基脱氧胆酸。在确定其细菌来源与催化机制后,本研究发现3-乙酰基脱氧胆酸在人群中广泛存在,并可调控肠道微生物互作。综上所述,本研究从酶学视角为微生物胆汁酸与宿主的关系提供了新的见解。
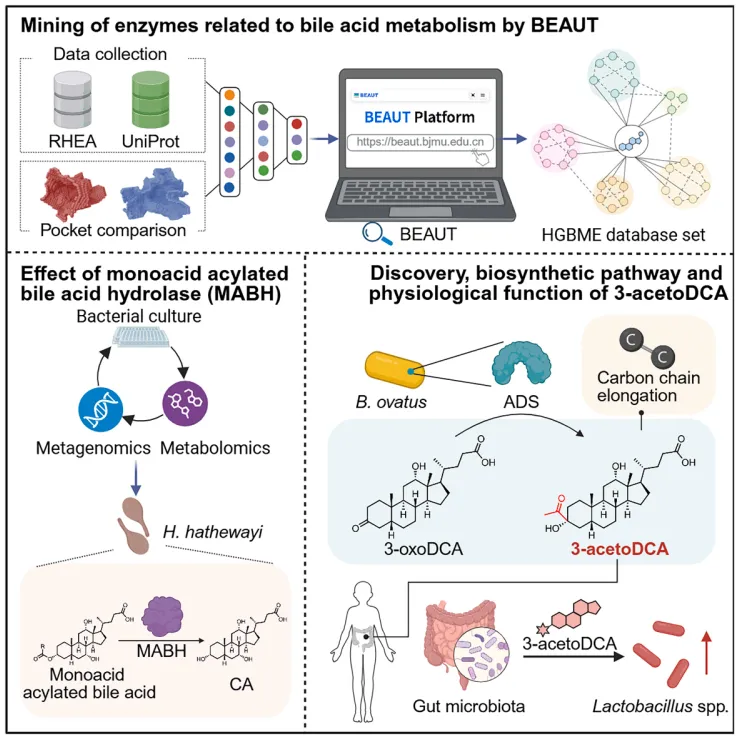
论文ID
实验设计
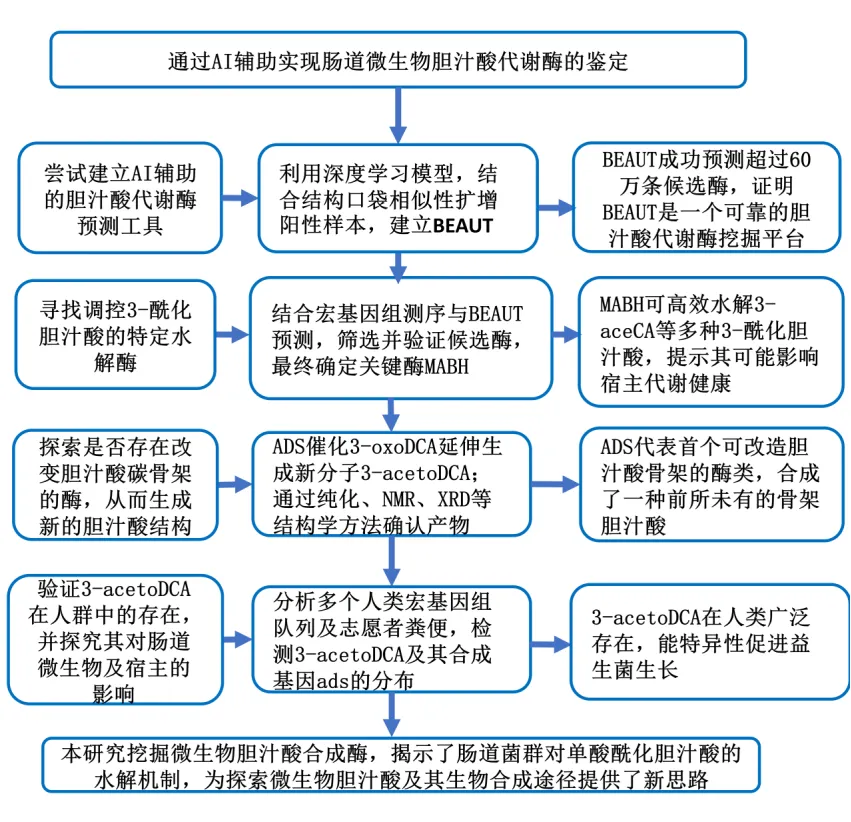
结果
1.BEAUT平台的开发
BEAUT利用深度学习技术预测未被探索的胆汁酸代谢酶。研究者采用ESM-2模型生成蛋白质序列的功 能感知表征,并通过密集神经网络传递这些方法,以预测给定序列是否编码胆汁酸代谢酶(图1A-1C、S1A和S1B)。该模型仅以酶的序列作为输入,进而评估序列能否利用胆汁酸作为底物。研究者将酶编号预测作为筛选过程的附加维度。这种多维筛选策略提升了对数据不足的耐受性,并在筛选大规模序列数据集时实现了可接受的性能表现。

图1. BEAUT的模型开发流程。(A) BEAUT工作流程示意图。在“数据收集与模型开发”环节中,从RHEA数据库和UniProt数据库收集了阳性序列(与BAs发生反应的酶)与阴性序列(不与BAs发生反应的酶)。筛选扩增后的阳性序列,使用ESM-2模型对蛋白质序列进行功能感知表征编码,并通过密集神经网络传递这些表征,以预测肠道微生物组序列是否为BA代谢酶。随后在菌株和酶水平上对BEAUT模型进行了验证。通过酶活性测定实验对预测的BA相关候选酶进行了验证,本研究最终确认了两种BA代谢酶及其介导的反应过程。(B) 扩增序列收集示意图。从具有高度多样化BA转化能力的肠道微生物中,筛选出与初始阳性序列具有高结合口袋相似性的扩增序列。(C) BEAUT模型训练集、验证集和测试集的构建流程。在选定阳性测试样本,将剩余阳性样本与随机选取的阴性样本分别划分为5类进行交叉验证。(D) BEAUT挖掘的BA相关候选酶的酶分类预测,该分类通过CLEAN工具完成。 (E) BEAUT挖掘的BA相关候选酶的前10位酶分类,其EC编号由CLEAN工具预测。(F) BEAUT挖掘的BA相关候选酶在门、纲、目、科、属、种级别的分类分布。
研究者在人工筛选的反应知识库子集中收集以胆汁酸为底物的代谢反应。通过UniProt数据库获取标注这些反应的酶序列作为正样本,同时将标注其他RHEA反应的序列作为负样本。此外,研究者还从最新文献中手动补充了若干序列作为正样本。最终共收集到1,032个正样本和108,319个负样本(图S1A)。在去除片段序列及90%序列同一性水平的冗余序列后,保留151条独特正序列,并将其确定为核心正样本(表S1)。
随后,研究者根据序列长度筛选阴性样本以匹配阳性样本的分布,最终获得102,403条阴性序列。这种不平衡的正负样本数据导致无法训练有效的机器学习模型,因此必须进行数据增强。研究者首先分析了来自蛋白质数据库的8个酶-胆汁酸复合物结构,使用Cavity程序提取这些蛋白质中的结合口袋,并根据共同骨架结构对齐胆汁酸配体。研究者对这些结构的分析(数据S1)表明:胆汁酸分子具有相对相似的构象,且倾向于结合体积大于1,000立方埃的结合口袋。随后,研究者使用PocketMatch计算了这些蛋白质复合物中胆汁酸结合口袋的两两相似性,结果表明尽管序列和结构相似性较低,这些蛋白质可能具有较高的口袋相似性(数据S1)。例如,磺基转移酶与另外两种胆汁盐水解酶在序列、结构和功能上存在差异,但存在较高的口袋相似性。这些数据表明具有与初始阳性样本相似底物结合口袋的酶可能同样属于胆汁酸酶。为验证这一观点,研究者进一步收集了初始阳性样本的蛋白质结构,并使用Cavity程序提取其结合口袋(表S1)。以整体蛋白质结构质量及结合口袋特征为筛选依据,本研究最终生成了85个参考口袋。通过口袋两两相似性比较,研究者发现以胆汁酸为底物的酶类比非底物酶类具有更高的口袋相似度(图S1C)。该结果进一步验证了本研究的假设:通过口袋相似性分析可实现阳性数据的扩增。
研究者依据先前的研究报告,选择了7株具有高多样性及强胆汁酸代谢能力的肠道菌基因组作为扩增样本来源。通过流程I对所选7株菌的蛋白质序列进行筛选,剔除与任何存在50%以上一致性的阴性样本序列。随后,研究者对剩余蛋白质进行结构预测,并提取与初始阳性样本体积相当的潜在结合口袋。以整体蛋白质结构质量、口袋质量为筛选依据,最终本研究获得10,867个口袋。研究者使用PocketMatch计算查询口袋与初始阳性样本参考口袋的相似度(图1B与S1B),筛选出口袋相似度大于0.7的序列用于后续扩增。将这些序列与151个初始阳性样本合并,并在90%序列相似度水平进行去重后,最终获得2,472个扩增阳性样本用于构建BEAUT模型(表S1)。
研究者采用交叉验证方法,利用增强后的阳性样本与阴性序列训练了五个模型(图1C)。模型训练过程中的训练损失与验证损失分析表明未出现过拟合现象(图S1D)。在平衡测试集上对五个模型进行评估,其AUPRC平均值为0.80,F1分数平均值为0.72,召回率平均值为0.75。完整评估指标列表参见表S2。研究者选择在该平衡测试集上AUPRC表现最佳的模型作为最终BEAUT模型。
运用BEAUT模型,研究者对人类微生物组计划参考基因组进行了基于序列的虚拟酶筛选。共收集到2,340,761条处于阳性序列长度范围内的蛋白质序列进行筛选。研究者对预测阳性序列进行进一步注释,并通过流程II进行筛选。BEAUT模型预测出614,616条序列为候选胆汁酸代谢酶(图S1E),随后采用EFI工具将序列聚类为118,599个簇。研究者进一步使用CLEAN进行EC编号预测以评估潜在反应类型,发现阳性序列分布于全部7大类酶中,呈现功能多样性(图1D与1E)。在门分类水平上,拟杆菌门被预测含有最丰富的胆汁酸代谢酶(图1F)。在属分类水平上,有两类细菌显示出更高的胆汁酸代谢酶丰度,且不同菌株间的预测阳性序列数量变异极小(图1F)。所有预测的胆汁酸代谢酶已整合为人类广义微生物胆汁酸代谢酶(HGBME)数据集,并通过本研究的网络服务器(https://beaut.bjmu.edu.cn)公开共享。
2. BEAUT模型在菌株与酶水平上的评估
研究者通过实验验证BEAUT模型在菌株和酶水平上的预测准确性(图2A)。在菌株层面,研究者将108株肠道菌株与5种胆汁酸底物共培养,包括CA、CDCA、DCA、LCA和3-oxoDCA。经过48小时孵育后,研究者采用液相色谱-质谱联用技术分析了胆汁酸代谢谱。胆汁酸底物的减少量可作为肠道菌株胆汁酸代谢能力的指标(图2B)。研究发现,通过BEAUT模型预测的胆汁酸代谢酶数量与实测的菌株胆汁酸代谢能力呈正相关关系(图2C和图S2A)。
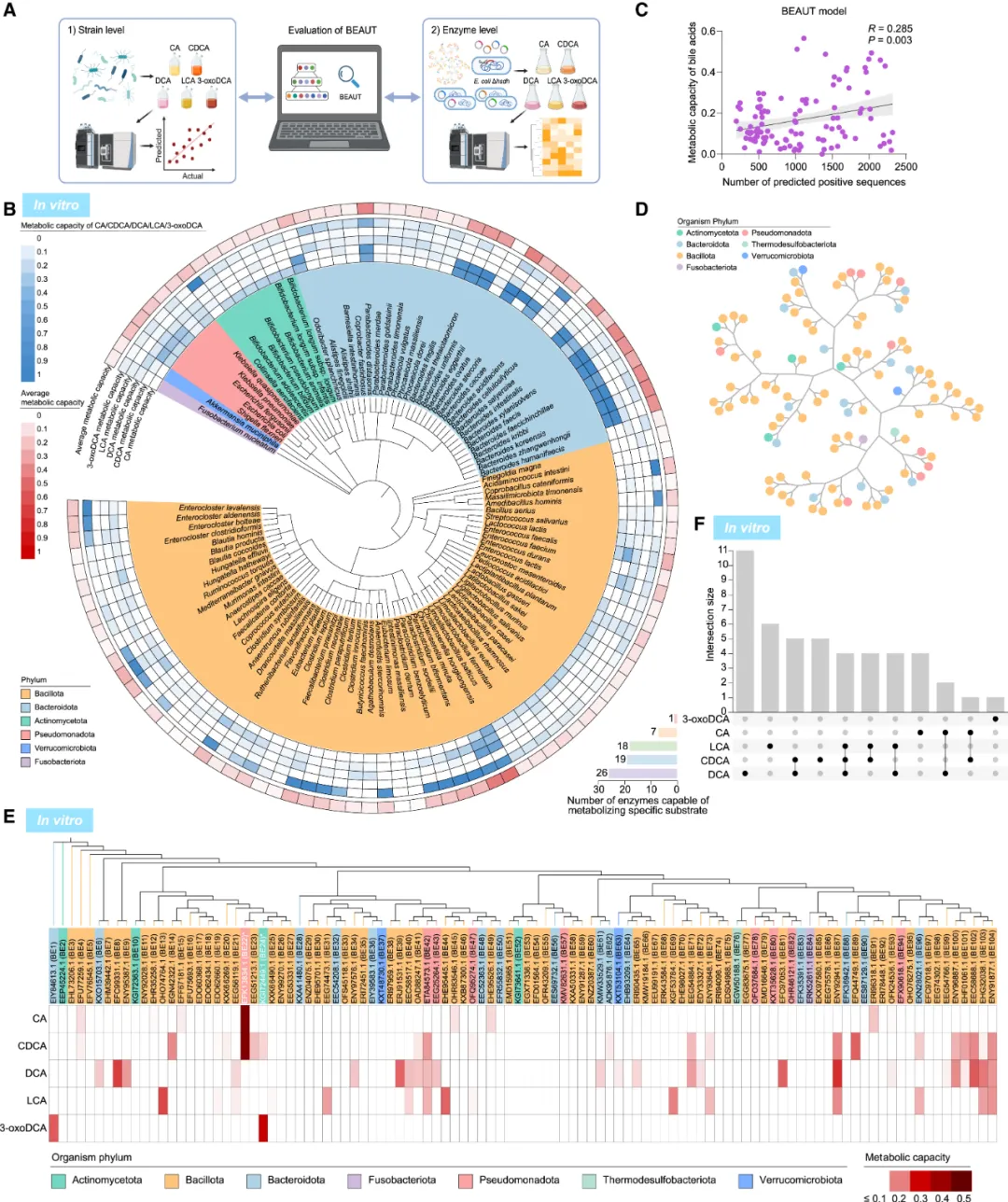
图2.BEAUT模型在菌株和酶水平的评估。(A) 菌株与酶模型验证示意图。(B) 108种肠道微生物的系统发育树及其胆汁酸代谢能力(体外实验)。 (C) 胆汁酸体外代谢能力与BEAUT预测阳性序列数量的相关性分析。(D) 验证所用酶的系统发育树。酶按来源菌株的门类着色。(E) 热图显示验证酶对不同胆汁酸底物的体外代谢能力。 (F) UpSet图展示经体外实验验证的不同底物阳性胆汁酸代谢酶。
3. 肠道菌群中单酸3-酰化胆汁酸水解酶的发现
既往研究已发现胆汁酸存在酰化修饰,包括单酸酰化与二酸琥珀酰化。本研究近期证实3-琥珀酰化胆酸是由琥珀酰基胆汁酸酰基合成酶催化合成。值得注意的是,BEAUT模型在未经过相应阳性样本训练的情况下,成功溯源到BAS-suc酶(图3A)。研究者对该BAS-suc蛋白簇的进一步验证揭示了多个此前未知的BAS-suc蛋白(图3B与3C),这些蛋白与BE105类似,均能催化胆汁酸与琥珀酸之间的反应(图S3A)。这些BAS-suc酶对不同胆汁酸表现出广谱性底物选择性(图S3A)。
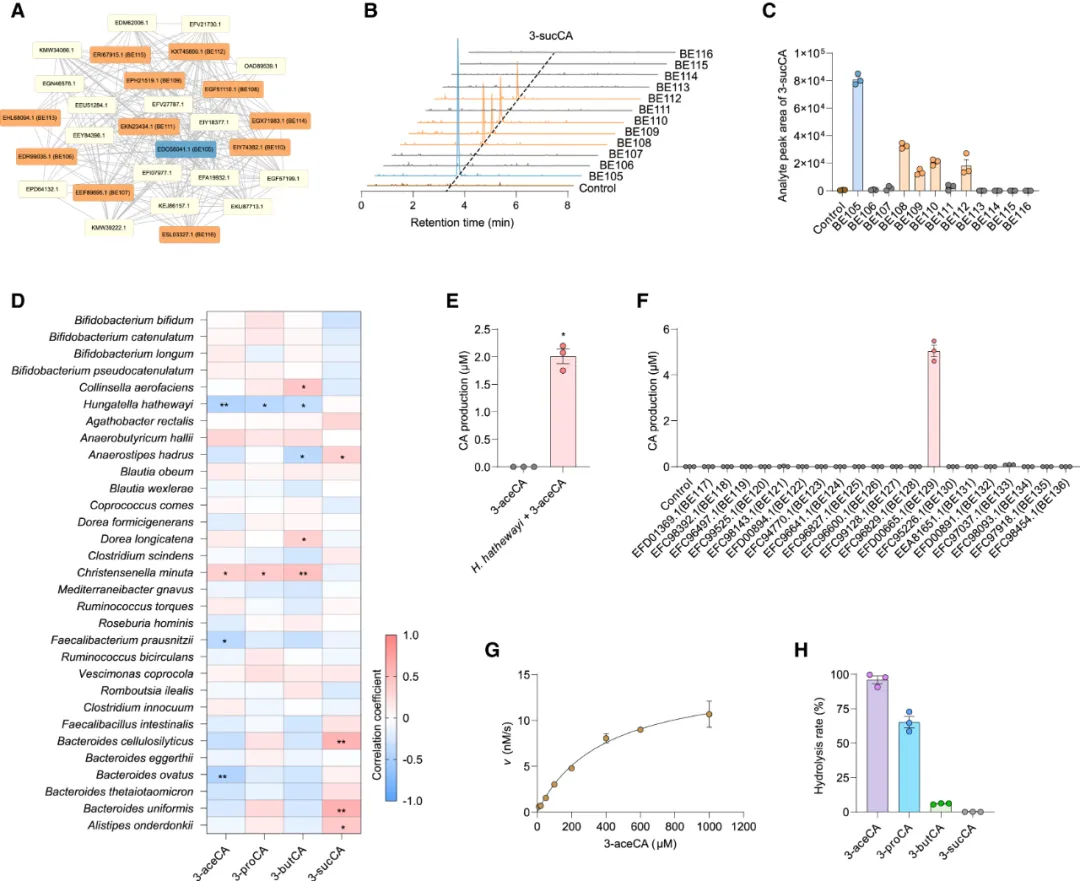
图3. MABH负责3-aceCA的水解作用。(A) 通过EFI-EST从HGBME数据库中生成的潜在BAS-suc蛋白簇。 (B和C) 不同大肠杆菌菌株裂解液催化酶促反应产生的离子色谱图(B)及定量产量(C)。(D) 热图显示30名健康志愿者粪便样本中酰化胆汁酸水平与特定肠道菌种的相关性。 (E) H. hathewayi水解3-aceCA生成CA的定量产量。 (F) BEAUT预测的3-aceCA水解活性。(G) HhMABH(BE129)的米氏动力学曲线。 (H) HhMABH与不同3-酰化CA孵育的水解速率。
研究者尝试利用BEAUT探索3-酰化胆汁酸的潜在调控通路,因此招募30名志愿者并对其粪便样本进行宏基因组测序与靶向胆汁酸分析。研究发现,单酸型3-酰化胆汁酸的浓度与特定肠道共生菌丰度呈显著负相关(图3D),提示肠道微生物群中存在潜在的3-酰化胆汁酸水解通路。研究者对相关性最强的Hungatella hathewayi菌株进行体外孵育实验,发现该菌能高效降解3-aceCA(图3E)。随后,研究者采用BEAUT预测Hungatella hathewayi菌株中潜在的胆汁酸代谢酶,通过CLEAN工具对预测候选酶进行功能注释,并筛选出分类为羧酸酯水解酶(EC 3.1.1)的前20个候选酶,并对其进行了深入的分析(图S3B、S3C)。通过候选酶的过表达与活性验证,最终确定功能酶为负责水解3-aceCA的关键酶,将其命名为HhMABH(图3F)。经纯化HhMABH蛋白(图S3D)并进行动力学分析,发现该酶对3-aceCA的米氏常数为0.39 mM(图3G)。通过对多种3-酰基胆酸的底物适应性分析,研究者进一步发现HhMABH能够水解3-aceCA、3-proCA和3-butCA,尤其对前两种底物表现出较高活性,但对3-sucCA无催化活性(图3H和图S3E)。
4. ADS催化形成先前未经表征的骨架结构BA 3-乙酰氧基脱氧胆酸
细菌对胆汁酸的修饰主要发生在活性官能团上,例如羟基异构化、脱羟基化、酰基化和羧基酰胺化,而非涉及碳骨架的改变。然而,碳骨架的修饰可能导致分子生物活性的显著差异。尽管如此,迄今为止尚未发现存在替代骨架胆汁酸。在实验验证结果中,研究者筛选到一个可能参与碳链延伸的硫胺素焦磷酸依赖性酶,且该酶至今在胆汁酸代谢中尚未见报道。此外,研究者过表达了BE1相关基因簇中的11种酶,发现大多数酶能以不同程度消耗3-氧代脱氧胆酸,但对其他胆汁酸底物影响甚微(图4A和4B)。尽管3-氧代脱氧胆酸水平降低,本研究并未观察到已知3-氧代脱氧胆酸衍生物(如脱氧胆酸)的增加(图S4A)。
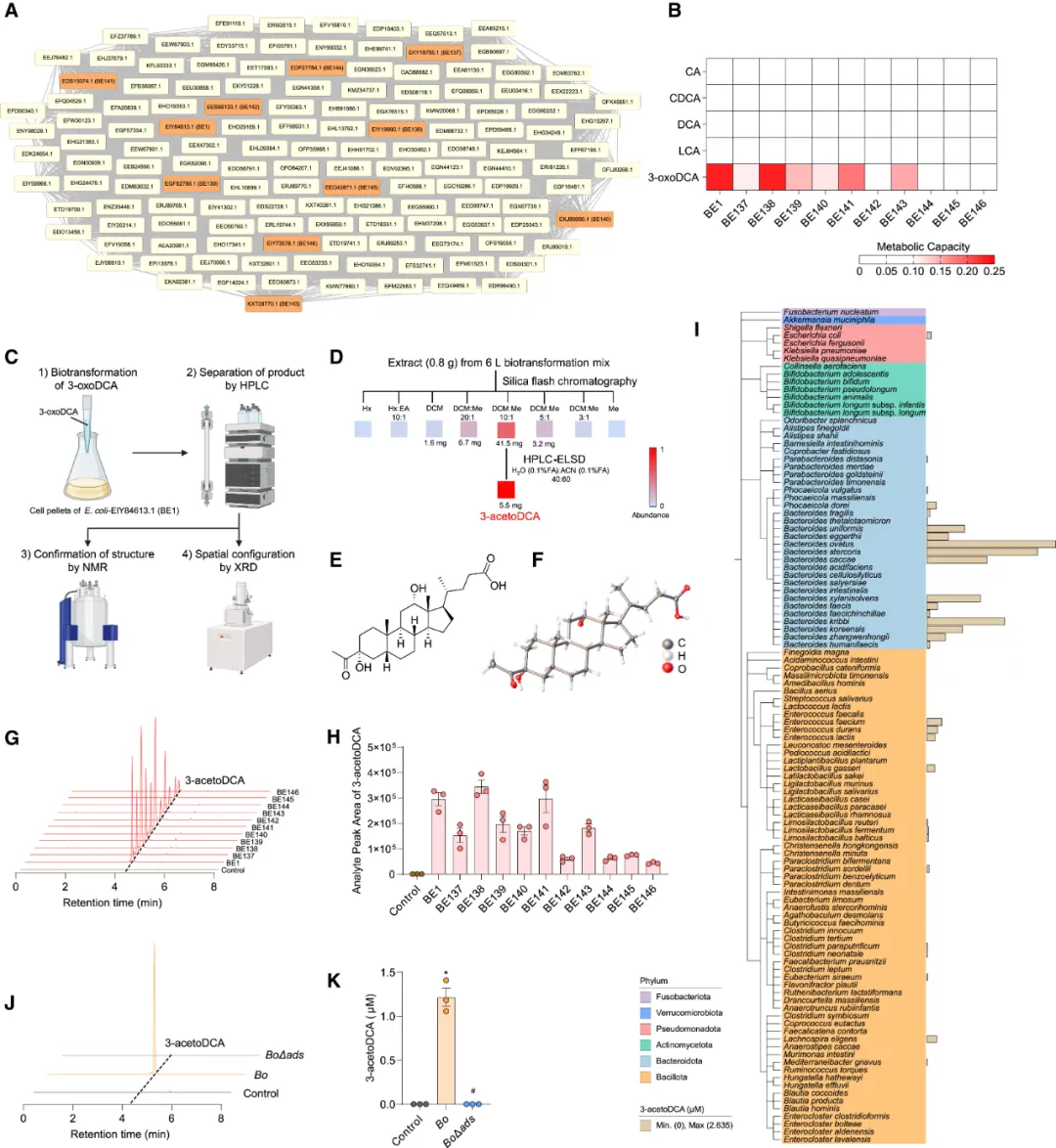
图4. ADS催化胆汁酸3-乙酰氧基脱氧胆酸的形成。(A)通过EFI-EST工具从HGBME数据库中生成的BE-1相关基因簇(B)热图显示选自BE-1相关基因簇的潜在3-氧代脱氧胆酸代谢酶对不同胆汁酸底物的代谢能力。(C)未报道骨架胆汁酸3-乙酰氧基脱氧胆酸的分离鉴定流程图。(D)大肠杆菌转化3-氧代脱氧胆酸后的组分分离流程。(E)3-乙酰氧基脱氧胆酸的化学结构式。(F)通过X射线晶体学分析获得的3-乙酰氧基脱氧胆酸热椭球图。(G和H)不同大肠杆菌菌株催化产生的3-乙酰氧基脱氧胆酸代表性提取离子流色谱图(G)及定量产量(H)。(I)108种肠道微生物的系统发育树及其3-乙酰氧基脱氧胆酸产量。 (J和K) 培养基对照组、培养组及缺陷菌株组产生的3-乙酰氧基脱氧胆酸的离子色谱图(J)及其定量产量(K)。图(K)中p值采用Kruskal-Wallis检验结合Dunn事后检验测定。
在研究BE1酶对3-oxoDCA转化产物的过程中,研究者注意到反应后出现了一个m/z为433.2957的LC-MS峰(图S4B)。通过连续多重色谱及联用技术,结合基于LC-MS的分子量追踪,研究者获得了具有目标分子量的纯化合物,并采用NMR解析了其分子结构(图4C、4D)。结果表明底物3-oxoDCA的3位碳原子被延长了一个乙基酮基团(图4E、S4C–S4I)。进一步通过单晶X射线衍射测定其三维结构,最终确认为3-acetoDCA(图4F)。
研究者对酶BE1相关簇中的11种酶进行了重新分析,发现所有这些酶均能产生3-acetoDCA,并将该酶簇统称为ADS(图4G和4H)。在使用不同培养基的条件下,研究者在健康志愿者粪便来源的离体群落(SECs)中观察到了3-acetoDCA的产生(图S4J)。此外,研究者从实验室肠道菌株库的108个物种中筛选了3-acetoDCA的细菌来源。研究发现Bacteroides ovatus的3-acetoDCA产量最高(图4I),且不同Bacteroides ovatus菌株均能产生3-acetoDCA(图S4K和S4L)。通过同源比对,研究者在Bacteroides ovatusATCC 8483的基因组中鉴定出ADS同源蛋白EDO13458.1(BE147),并通过同源重组使Boads基因失活,构建了靶向基因组突变株(图S4M和S4N)。与野生型卵形拟杆菌ATCC 8483相比,BoΔads菌株完全丧失了产生3-acetoDCA的能力(图4J和4K),这表明BoADS是卵形拟杆菌中负责合成3-acetoDCA的关键酶。
3-acetoDCA的分子结构不同于传统胆汁酸(图S4O-S4Q)。除强极性的羧酸侧链外,3-acetoDCA在其四环结构的疏水性甾体骨架部分还具有一个额外的乙基酮侧链(图S4Q)。近期研究通过非靶向代谢组学分析,以m/z 319.24和m/z 337.25的通用骨架离子作为胆汁酸诊断性碎片,鉴定出多种胆汁酸。研究者推测3-acetoDCA可能因骨架延伸而呈现特殊分子特征。非靶向代谢组学数据证实,3-acetoDCA未显示传统胆汁酸的特征离子,而是呈现特殊的433.2957 m/z峰(图S4O与S4P)。基于当前发现,未来代谢组学工作流程的优化将有助于鉴定更多胆汁酸。
5. BoADS的晶体结构与催化机制
BoADS是一种依赖ThDP的酶,属于DXPS家族。该酶亚家族催化丙酮酸与d-GAP反应生成DXP,该反应是维生素B1和B6生物合成中的关键限速步骤。相比之下,作为胆汁酸代谢中未被报道的酶,BoADS代表了以胆汁酸为底物的特殊ThDP依赖性酶(图5A–5C和图S5A–S5D)。在最佳pH和温度条件下进行的动力学分析表明,BoADS的米氏常数为95.03 μM(图5C、图S5B和图S5C)。
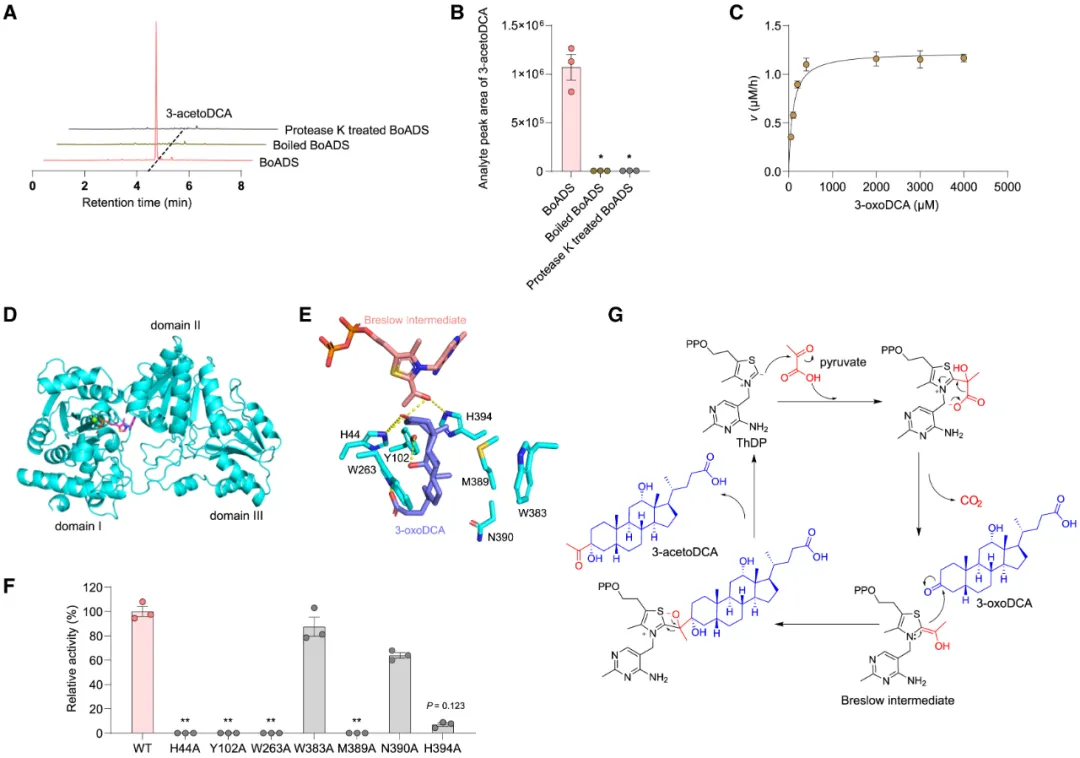
图5. BoADS-ThDP的晶体结构及其催化机制。(A和B)不同条件下BoADS蛋白酶促反应中3-乙酰氧基脱氧胆酸的代表性提取离子流色谱图(A)与定量产物生成量(B)。(C)以不同浓度3-氧代脱氧胆酸为底物时BoADS的米氏曲线。(D)BoADS-ThDP整体结构由I、II、III三个结构域组成。ThDP以洋红色棒状模型显示,Mg²⁺离子以绿色球体表示。(E)BoADS与Breslow中间体及3-氧代脱氧胆酸的复合物模拟结构。氢键以黄色虚线标示。(F)BoADS及其变体与3-氧代脱氧胆酸结合位点相关的相对催化活性。(G)BoADS的催化机制推测。
为阐明BoADS的催化机制,研究者解析了其与ThDP复合物的晶体结构,分辨率为2.42 Å,每个不对称单元包含两个分子(表S4)。BoADS-ThDP的整体结构呈现出与DXPSs相似的结构折叠模式,并包含三个独立结构域(I、II和III)(图5D)。每个结构域均由中央五股或六股平行β-折叠及数量不等的环绕α-螺旋构成。研究发现ThDP结合口袋位于结构域I和II的界面处:ThDP的二磷酸基团与Mg²⁺离子共同与残基H75、D147、N174、N176、K247和H262发生相互作用(图S5E);ThDP的嘧啶环与F358形成π–π堆叠作用,并与G116和E333形成氢键(图S5E)。对上述残基进行点突变后均观察到催化活性显著降低或完全丧失,证实这些残基在ThDP结合中起关键作用(图S5F)。
在BoADS的催化过程中,辅因子ThDP首先与底物丙酮酸反应形成Breslow中间体,该中间体随后与另一底物3-oxoDCA发生反应。为探究底物结合模式,研究者将Breslow中间体与3-oxoDCA共同建模至BoADS活性位点。研究发现Breslow中间体的羟基与H44和H394形成氢键,而3-oxoDCA的C3-羰基、C12-羟基和C24-羧基分别与H44、Y102和W263形成氢键(图5E)。更重要的是,本研究发现3-oxoDCA的烃环与W263和M389形成了”夹心式”疏水相互作用,其中W263位于一侧,M389位于另一侧(图5E)。H44A、H394A、Y102A、W263A及M389A酶变体均显示出催化活性下降或丧失,证实了这些残基在底物结合中的关键作用(图5F)。
通过DALI结构比对分析,研究者发现BoADS与DrDXPS具有最高的结构相似性,其Cα原子的RMSD为1.19。BoADS与DrDXPS的底物存在差异:BoADS的底物为3-oxoDCA,而DrDXPS的底物为d-GAP(图S5G)。在结构层面,BoADS呈现出与DrDXPS相似的结构折叠方式(图S5H与S5I),但两者具有不同的底物结合口袋,从而导致其底物识别活性存在差异(图S5J与S5K)。此外在DrDXPS中,关键残基W263和M389被替换为G305和G428,这一关键残基在BoADS中会与3-oxoDCA形成相互作用,这一发现突显了BoADS与3-oxoDCA结合的特殊模式。
基于上述晶体结构和定点突变分析结果,研究者提出了BoADS的催化机制(图5G)。具体而言:ThDP辅因子紧密结合于BoADS结构域I与II之间,攻击丙酮酸的α-酮基,经脱羧反应生成Breslow中间体;该中间体继而攻击3-oxoDCA的3-酮基,最终通过ThDP的离去生成3-乙酰氧基DCA。这一机制深化了本研究对自然界利用酶促反应实现次级胆汁酸库多样化的认知。
6.3-acetoDCA在人类群体中广泛存在,对肠道菌群具有特殊影响
本研究随后探究了3-acetoDCA是否天然存在于人类肠道中。通过分析来自公共数据库中三个独立人类队列的宏基因组数据,研究者发现Boads基因在不同地区广泛分布(图6A–6C)。通过进一步代谢组学分析,研究者在健康志愿者粪便样本中证实了3-acetoDCA的存在(图6D)。同时,对志愿者粪便的宏基因组分析表明,3-acetoDCA的浓度与Boads基因及卵形拟杆菌的丰度呈正相关(图6E和6F)。
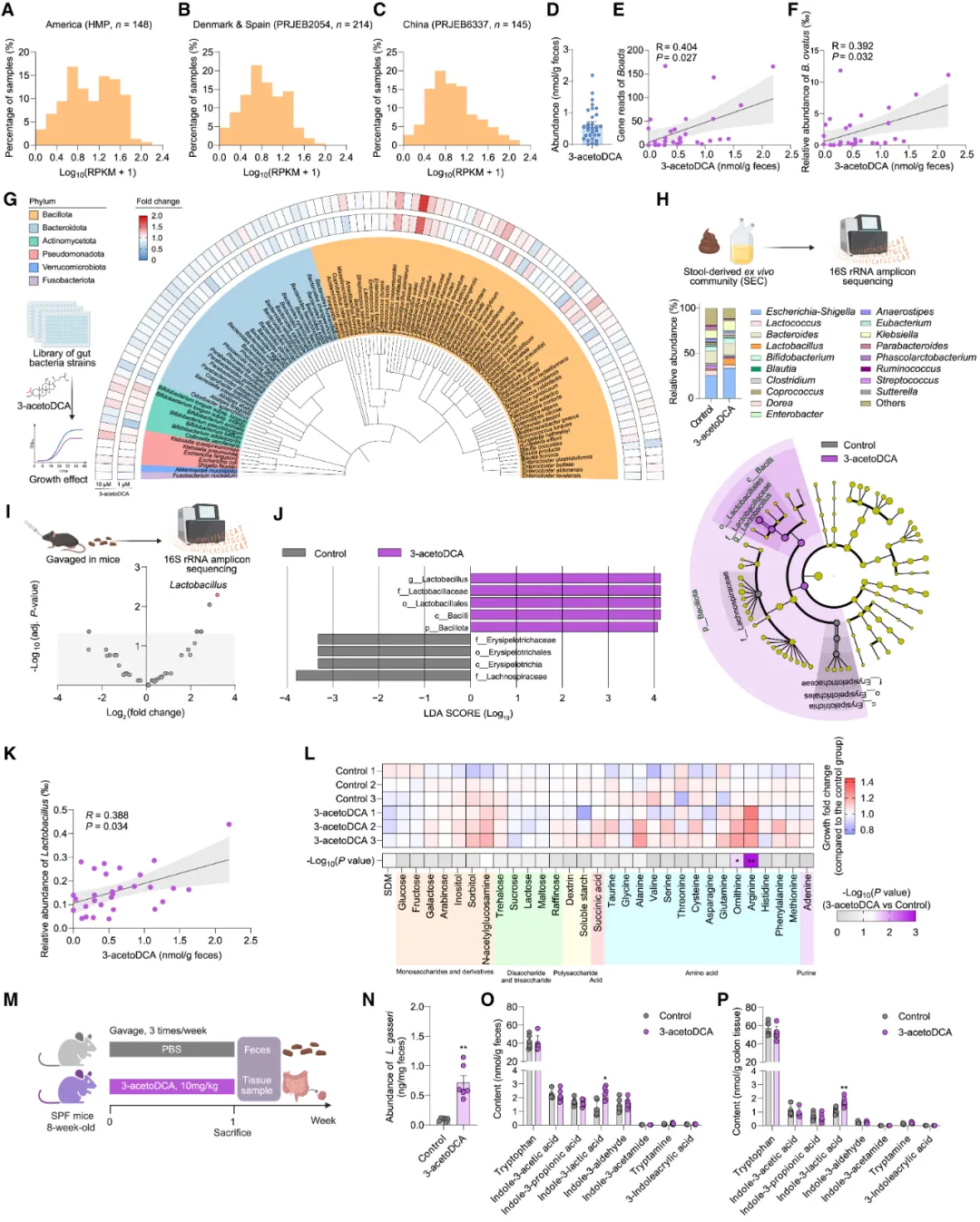
图6.3-乙酰氧基脱氧胆酸在人体内广泛存在并促进乳酸杆菌增殖。(A-C)卵形拟杆菌的3-乙酰氧基DCA合成基因在三大数据库中广泛存在:HMP(A)、PRJEB2054(B)和PRJEB6337(C)。(D)30名健康志愿者粪便中3-乙酰氧基DCA的水平。(E-F)3-乙酰氧基DCA与基因Boads(E)及卵形拟杆菌丰度(F)的相关性分析。(G)108种肠道微生物的系统发育树及不同浓度3-乙酰氧基在单个菌株水平对肠道细菌生长的影响。(H)在添加10μM 3-乙酰氧基DCA或PBS对照的mGAM培养基中厌氧培养48小时后,人源粪便体外群落的组成。(I-J)SPF小鼠每周3次接受PBS(对照)或10mg/kg 3-乙酰氧基DCA处理,持续1周。收集终点粪便样本进行16S rRNA扩增子测序。(I)对照组与3-乙酰氧基DCA组16S rRNA基因测序数据的火山图。 (J) 基于LEfSe分析生成的分类学分支图。 (K) 30名健康志愿者体内3-乙酰氧基脱氧胆酸与乳酸杆菌丰度的相关性分析。 (L) 在添加不同营养物质(0.5 mg/mL)的半限定培养基中,于3-乙酰氧基脱氧胆酸或DMSO(对照组)条件下培养加氏乳杆菌。 (M-P) 如图示方案(M)所示,对照组与3-乙酰氧基脱氧胆酸处理的SPF小鼠粪便中加氏乳杆菌丰度(N)、结肠内色氨酸代谢物水平(O)及结肠内容物色氨酸代谢物水平(P)。
研究者进一步检测了3-acetoDCA对传统胆汁酸受体的作用,通过荧光素酶报告基因检测发现,该化合物对FXR受体、VDR受体、PXR受体及TGR5受体的活性均无影响(图S6A-S6D)。考虑到某些胆汁酸可通过调节肠道菌群组成来影响宿主功能,研究者继而探究了3-acetoDCA对肠道微生物群落生长的影响,发现该物质可促进乳酸杆菌属的生长(图6G-6J,S6E-S6N)。具体而言,3-acetoDCA在体外能以剂量依赖的方式特异性促进乳酸杆菌属的生长(图6G),而其他已被深入研究的胆汁酸(如CA、CDCA、DCA、LCA和3-oxoDCA)均未显示出对乳酸杆菌属生长的促进作用(图S6E)。与此形成对比的是,不同菌株的3-acetoDCA处理下均表现出相似的生长促进效应(图S6H-S6J)。通过对多种乳酸杆菌属菌种的表征,研究者再次证实3-acetoDCA可促进乳酸杆菌属菌种的生长(图S6K-S6N)。在标准化菌群培养体系和小鼠模型中,3-acetoDCA处理亦能显著提高离体与体内条件下乳酸杆菌的丰度(图6H-6J,S6F,S6G)。此外,研究者在粪便样本中观察到3-acetoDCA浓度与乳酸杆菌丰度间存在显著正相关性(图6K)。
接下来,研究者尝试探究3-乙酰基-DCA促进加氏乳杆菌生长的作用机制。通过在半限定培养基中添加不同营养物质(包括单糖、双糖、多糖及氨基酸)并观察3-乙酰基-DCA处理与否对加氏乳杆菌生长的影响,研究者发现当不添加外源营养物质时,3-乙酰基-DCA几乎不表现出促生长作用。值得注意的是,当补充精氨酸或鸟氨酸后,经3-乙酰基-DCA处理的加氏乳杆菌生长显著改善(图6L、S6O和S6P)。既往研究报道表明,精氨酸是乳酸杆菌生长的必需营养素,其缺失会抑制乳酸杆菌的生长。
考虑到既往研究报道Lactobacillus属细菌能产生多种具有改善代谢性疾病潜力的色氨酸衍生物,研究者进一步评估了3-乙酰氧基脱氧胆酸(3-acetoDCA)在体内处理下色氨酸衍生物的水平(图6M)。通过qPCR验证发现,灌胃给予3-acetoDCA可显著提高小鼠体内L. gasseri的丰度(图6N),同时显著增加粪便和结肠组织中吲哚-3-乳酸浓度(图6O和6P)。
讨论
代谢物充当肠道菌群与宿主间的信使。作为宿主与肠道菌群的共代谢产物,胆汁酸在脂质代谢、免疫调节和细菌感染中发挥关键作用。肠道菌群可利用宿主源性胆汁酸产生多种微生物胆汁酸,包括DCA、LCA、氨基酸结合型胆汁酸、3-酰化胆汁酸、氧化型胆汁酸(3-氧代、6-氧代、7-氧代和12-氧代)等。不同结构的胆汁酸可通过激活特定受体或影响特定肠道菌群的生长,进而调节宿主稳态与疾病。胆汁酸的微生物修饰作用构成调控生理过程的微生物密码。鉴定参与此类胆汁酸修饰的相关酶类,对于制定靶向策略、构建工程菌株以及理解胆汁酸修饰在健康与疾病中的作用至关重要。然而精准定位参与胆汁酸代谢的具体微生物酶仍是重大挑战。例如阐明从胆酸到脱氧胆酸的7α-脱羟基化途径耗时约40年。同样地,尽管早在宿主中发现氨基酸结合型胆汁酸由胆汁酸-辅酶A、氨基酸N-酰基转移酶合成,但直至近期才发现细菌胆汁酸水解酶/转移酶也能合成微生物结合型胆汁酸。在本研究中,研究者利用人工智能技术拓展了胆汁酸代谢酶的发现能力,通过开发BEAUT工作流程从大规模数据集中预测胆汁酸代谢酶。研究者验证了100余种候选酶,并鉴定出两种未被报道的酶,一种功能未表征的3-aceCA水解酶和一种新型骨架胆汁酸合成酶。为提升科研的可获得性,研究者开发了交互式网络服务器BEAUT(可通过https://beaut.bjmu.edu.cn访问)用于胆汁酸代谢酶预测。同时在该平台部署人类微生物组基因组中潜在胆汁酸代谢酶的HGBME数据集。用户可自由检索数据集并下载目标蛋白。
既往研究已发现胆汁酸存在单酸与双酸3-O-酰化修饰。研究者运用BEAUT技术,在未预先引入相关阳性序列的情况下,成功回溯到能催化琥珀酰化修饰的BAS-suc。目前尚不清楚3-酰化胆汁酸的组成是否受特定酰化胆汁酸水解酶的调控,这是一种类似于结合型胆汁酸受BSHs调控的机制。为鉴定特定胆汁酸代谢酶,研究者开发了基于BEAUT的系统化研究流程:(1)采用组学技术筛选与特定胆汁酸代谢相关的菌株;(2)获取这些相关菌株并通过实验验证候选菌株能否将特定胆汁酸转化为预期产物;(3)利用BEAUT预测候选菌株基因组中所有与胆汁酸代谢相关的基因,随后根据预测评分和酶反应类型对潜在候选酶进行优先级排序,并验证其催化特定胆汁酸转化的能力。通过上述流程,研究者在H. hathewayi菌株中鉴定出一种先前未知的单酸酰化胆汁酸微生物水解酶MABH。已有研究表明3-酰化胆汁酸可通过肠道FXR-神经酰胺轴调控2型糖尿病。本研究的研究提示MABH可作为开发糖尿病疗法的潜在靶点,值得进一步开展临床研究。
胆汁酸分子的鉴定是一项复杂且耗时的工作。近期研究采用质谱与代谢组学技术,引入了MassQL算法等新策略,由此发现了未被报道的胆汁酸,包括多胺结合型胆汁酸以及新近发现的细菌胆汁酸酰胺化合物。BEAUT技术将海量测序数据与特定分子相关联,规避了传统的菌株分离和化合物纯化策略,从而加速了胆汁酸代谢酶的发现进程。肠道微生物群能够修饰胆汁酸的羟基、羧基等活性官能团,生成具有不同生物活性的胆汁酸,这些分子在免疫细胞分化和微生物调控中发挥重要作用。与常见的官能团修饰相比,改变胆汁酸碳-碳骨架的反应较为罕见且类型有限,但此类修饰往往能显著影响代谢物的活性。利用BEAUT微生物酶挖掘系统,本研究鉴定出一种微生物酶ADS,该酶能催化胆汁酸C3支链的形成,生成具有双支链结构的未记录骨架,代表了一类未被探索的双支链胆汁酸。通过对催化机制的深入分析,发现负责合成3-acetoDCA的BoADS酶属于DXPS家族。然而与初级代谢中其他成员的底物结合口袋相比,BoADS的口袋尺寸更大,并能与胆汁酸底物形成特殊的夹心式相互作用。对该口袋的后续分析将有助于胆汁酸代谢酶的发现与改造。
本研究的临床分析结果表明,Boads基因在不同人群中被广泛检出。此外,研究者发现3-acetoDCA能够促进益生菌乳杆菌属的生长,并提升吲哚衍生物的水平,这类物质因其潜在的代谢保护作用而被认知。后续研究应深入探究3-acetoDCA在生理与病理条件下的作用机制。
结论
本研究通过挖掘微生物胆汁酸合成酶,揭示了肠道菌群对单酸酰化胆汁酸的水解机制,由此阐明了肠道微生物如何通过对单酸酰化胆汁酸的精细调控维持葡萄糖稳态。更重要的是,本研究从功能未知的酶出发,发现了一种未被报道的广泛存在的长碳链骨架胆汁酸3-acetoDCA。该物质能特异性促进肠道共生菌的生长,富集小鼠体内的色氨酸代谢物,并在代谢性疾病中发挥潜在保护作用。本研究的发现为探索微生物胆汁酸及其生物合成途径提供了新思路,同时为发掘具有生理调节功能的胆汁酸提供了一种“模拟自然自上而下”的研究策略。
———-微科盟精彩文章———-
科研 | 内农大:代谢组学揭示肠道益生菌鼠李糖乳酪杆菌X9C17高产氨基酸的代谢特征(国人佳作)
科研 | 南京中医药大:甘草次酸通过Thbs1/PI3K-Akt/p53通路调节肠道菌群及其代谢物进而改善胃黏膜损伤
如果需要原文pdf,请扫描文末二维码



获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系微生态老师9,无需重复添加。
请关注下方名片,了解更多代谢组学知识