 夜雨聆风
夜雨聆风





群体遗传学分析脚本详解与学习文档
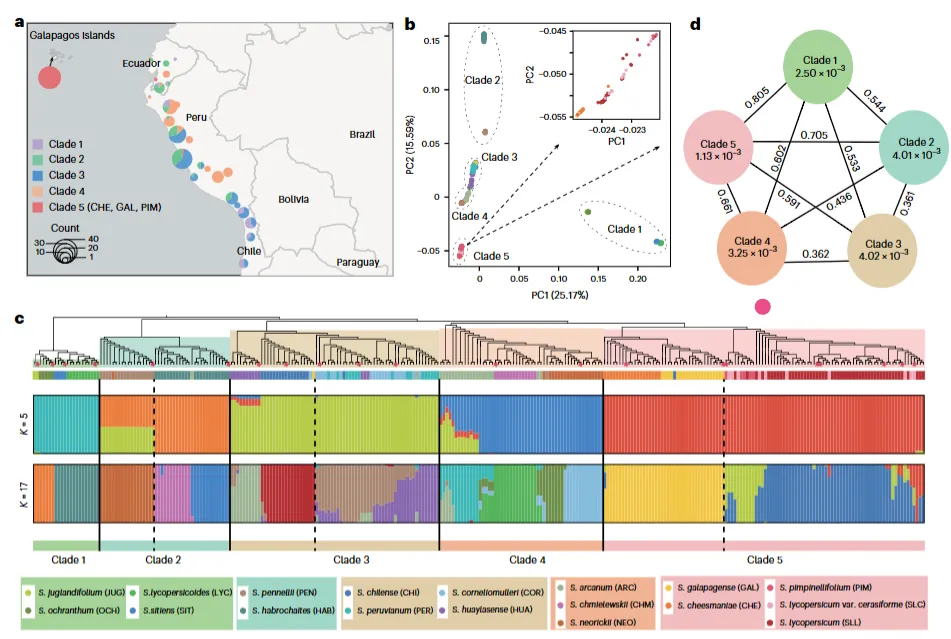
1. 环境与变量说明
脚本中使用了多个 Shell 变量(以 $ 开头),运行前需根据实际数据路径和文件名进行赋值:
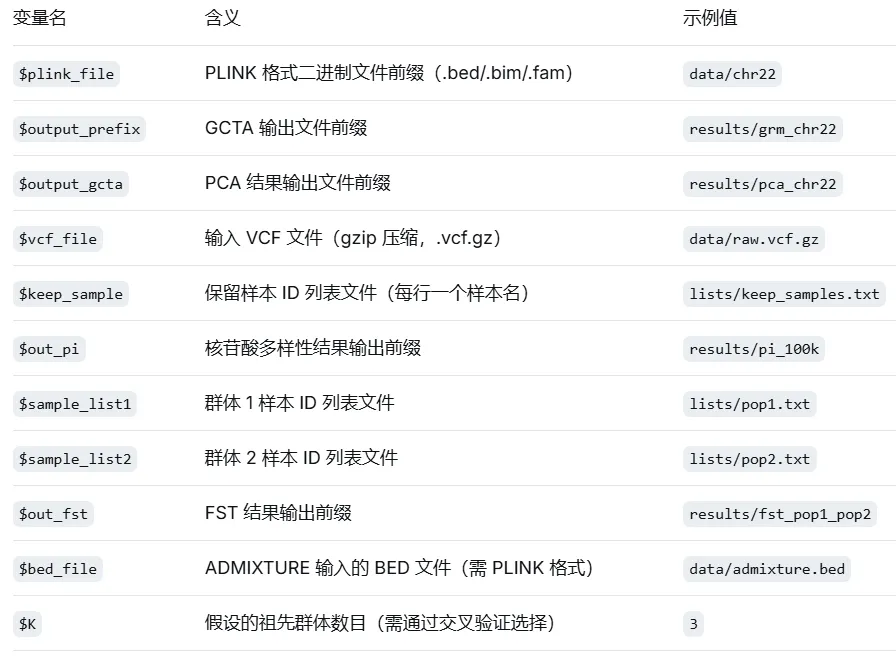
2. 遗传关系矩阵(GRM)与 PCA(使用 GCTA)
# 基于 PLINK 二进制文件计算遗传关系矩阵(GRM),仅使用常染色体,输出文件前缀为 $output_prefixgcta64 --bfile $plink_file --make-grm --autosome --out $output_prefix# 基于上一步计算得到的 GRM 进行主成分分析(PCA),提取前 10 个主成分gcta64 --grm $output_prefix --pca 10 --out $output_gcta
2.1 命令详解
-
gcta64:GCTA(Genome-wide Complex Trait Analysis)可执行文件,用于遗传分析。 -
--bfile $plink_file:指定 PLINK 二进制格式文件前缀(.bed, .bim, .fam)。 -
--make-grm:计算遗传关系矩阵(GRM),即所有样本对之间的亲缘系数(基于 SNP 信息)。 -
--autosome:仅使用常染色体 SNP(排除性染色体和线粒体)。 -
--out $output_prefix:输出文件前缀,生成$output_prefix.grm.bin,.grm.N.bin,.grm.id等文件。 -
--grm $output_prefix:指定上一步生成的 GRM 文件前缀。 -
--pca 10:对 GRM 进行特征分解,输出前 10 个主成分(PC1 ~ PC10)。 -
--out $output_gcta:PCA 结果输出前缀,生成$output_gcta.eigenval(特征值)和$output_gcta.eigenvec(特征向量,即每个样本在各 PC 上的坐标)。
2.2 输出文件解读

2.3 学习要点
-
GRM 用于估计样本间亲缘关系,可进一步用于遗传力估计、GWAS 校正等。
-
PCA 结果可用于检测群体分层、离群样本,并作为协变量纳入关联分析。
-
若样本量较大,计算 GRM 非常消耗内存和时间,可考虑使用
--grm-cutoff或--make-grm-gz(文本格式)调整。
3. 核苷酸多样性(π)计算(使用 VCFtools)
# 以 100 kb 为窗口、步长 10 kb 计算窗口内的平均核苷酸多样性(π),只保留指定样本vcftools --gzvcf $vcf_file --window-pi 100000 --window-pi-step 10000 --keep $keep_sample --out $out_pi
3.1 命令详解
-
vcftools:VCF 文件操作与统计工具。 -
--gzvcf $vcf_file:输入 gzip 压缩的 VCF 文件。 -
--window-pi 100000:滑动窗口大小(碱基数),此处为 100 kb。 -
--window-pi-step 10000:窗口步长(碱基数),即相邻窗口起点间隔 10 kb。 -
--keep $keep_sample:仅分析该文件中列出的样本(每行一个样本名)。 -
--out $out_pi:输出文件前缀,生成$out_pi.windowed.pi。
3.2 输出文件解读
$out_pi.windowed.pi 为制表符分隔的文本文件,包含以下列:
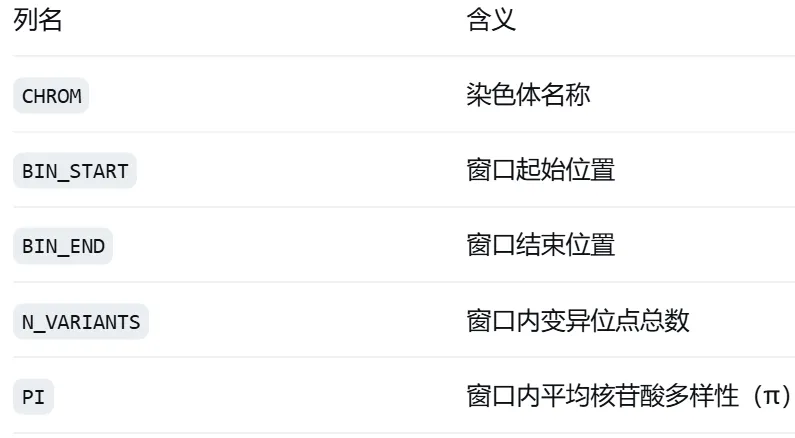
3.3 学习要点
-
π 衡量群体内遗传多样性,值越高表示多样性越丰富。
-
滑动窗口分析可识别基因组中多样性高/低的区域(如选择扫荡区域)。
-
注意:
--window-pi-step若不指定,则窗口不重叠(step = window size)。步长 < 窗口大小可实现重叠窗口。 -
若 VCF 文件很大,可先过滤 SNP(如 MAF > 0.05)以提高计算效率。
4. 群体分化指数(FST)分析(使用 VCFtools)
# 计算两个群体间的 Weir & Cockerham FST,以 100 kb 窗口、步长 10 kb 滑动分析vcftools --gzvcf $vcf_file --weir-fst-pop $sample_list1 --weir-fst-pop $sample_list2 --fst-window-size 100000 --fst-window-step 10000 --out $out_fst
4.1 命令详解
-
--weir-fst-pop $sample_list1:指定群体 1 的样本 ID 列表文件。 -
--weir-fst-pop $sample_list2:指定群体 2 的样本 ID 列表文件。 -
--fst-window-size 100000:滑动窗口大小(100 kb),在此窗口内计算平均 FST。 -
--fst-window-step 10000:窗口步长(10 kb)。 -
--out $out_fst:输出文件前缀,生成$out_fst.windowed.weir.fst。
4.2 输出文件解读
$out_fst.windowed.weir.fst 包含:
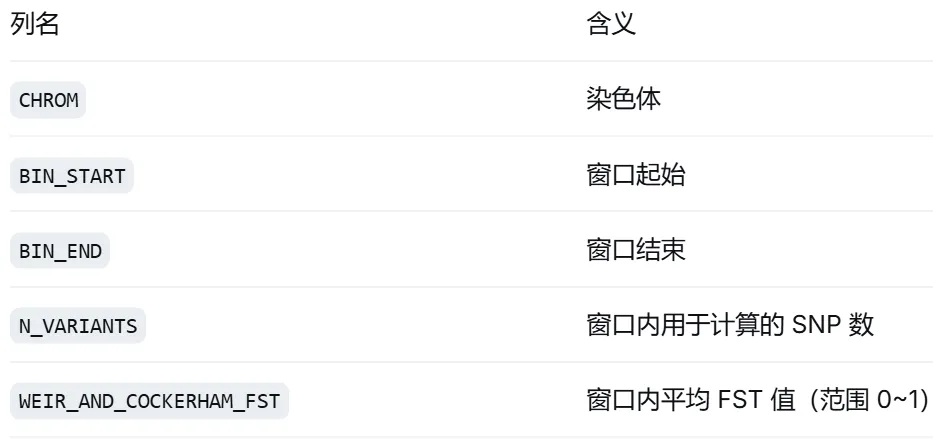
此外,VCFtools 还会输出基于每个 SNP 的 FST($out_fst.weir.fst)和日志文件。
4.3 学习要点
-
FST 衡量群体间遗传分化程度:0 表示无分化,1 表示完全分化。
-
高 FST 窗口可能受到选择作用(局部适应),低 FST 窗口可能为保守区域或基因流频繁区域。
-
通常需确保两个群体样本量足够(每个群体至少 5~10 个样本),且 SNP 经过质控(如 MAF > 0.05)。
5. 群体结构分析(使用 ADMIXTURE)
# 对给定的 BED 文件进行 ADMIXTURE 分析,假设 K 个祖先群体,执行 50 个并行线程,输出交叉验证错误率,同时将屏幕输出保存到 log.outadmixture --cv $bed_file -j50 $K | tee log.out
5.1 命令详解
-
admixture:基于 SNP 数据的模型聚类工具,估计每个个体的祖先成分比例。 -
--cv:进行 K 折交叉验证(默认 5 折),输出交叉验证错误率(CV error),用于选择最优 K 值。 -
$bed_file:PLINK 二进制 BED 文件前缀(需配套 .bim 和 .fam 文件)。 -
-j50:使用 50 个 CPU 线程并行计算(加速处理)。 -
$K:假设的祖先群体数目(通常需测试多个 K,如 2~10)。 -
| tee log.out:将标准输出同时显示在屏幕并保存到log.out文件中。
5.2 输出文件解读
ADMIXTURE 会生成以下文件(以 $bed_file 为基础名称,后缀添加 .K.Q 和 .K.P):
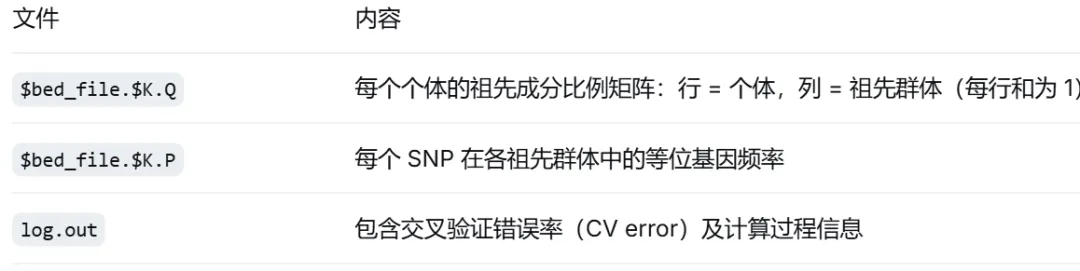
5.3 学习要点
-
确定最佳 K:运行多个 K 值(如 2~10),比较 CV error,选择最小或拐点对应的 K。
-
输入数据要求:SNP 应近似独立(通常先进行 LD 剪枝,如
--indep-pairwise 50 10 0.2)。 -
输出 Q 矩阵可用可视化作图(如
pong、pophelper或 R 包ggplot2)绘制群体结构堆叠柱状图。 -
并行数
-j需根据服务器资源调整,避免超限。
6. 完整工作流示例(含变量赋值)
#!/bin/bash# 设置变量plink_file="data/plink/chr22"output_prefix="results/grm/chr22"output_gcta="results/pca/chr22"vcf_file="data/vcf/chr22.vcf.gz"keep_sample="lists/kept_samples.txt"out_pi="results/pi/chr22"sample_list1="lists/pop_A.txt"sample_list2="lists/pop_B.txt"out_fst="results/fst/A_vs_B"bed_file="data/admixture/ld_pruned.bed"# 计算 GRMgcta64 --bfile $plink_file --make-grm --autosome --out $output_prefix# PCAgcta64 --grm $output_prefix --pca 10 --out $output_gcta# 核苷酸多样性vcftools --gzvcf $vcf_file --window-pi 100000 --window-pi-step 10000 --keep $keep_sample --out $out_pi# FSTvcftools --gzvcf $vcf_file --weir-fst-pop $sample_list1 --weir-fst-pop $sample_list2 \--fst-window-size 100000 --fst-window-step 10000 --out $out_fst# ADMIXTURE (测试 K=3)K=3admixture --cv $bed_file -j50 $K | tee log.k${K}.out
7. 注意事项与常见问题

8. 总结
本脚本涵盖了群体遗传学的核心分析方法:
-
GCTA:提供群体亲缘关系与 PCA 降维,是群体结构可视化和 GWAS 校正的基础。
-
VCFtools:高效计算 π 和 FST,分别揭示群体内多样性和群体间分化。
-
ADMIXTURE:基于模型的祖先成分推断,适用于探索群体混合历史。
建议按以下顺序实践:
-
准备 PLINK 二进制文件和 VCF 文件。
-
对数据进行质控和 LD 剪枝。
-
依次运行上述命令,检查输出文件格式。
-
使用 R 或 Python 绘制 PCA 散点图、π 与 FST 沿染色体分布图、ADMIXTURE 堆叠柱状图。
通过此脚本,可快速建立群体遗传学数据分析的基本流程。