夜雨聆风
夜雨聆风





长读长转录组从头组装工具综合评估,非模式生物研究有据可依

在生命科学领域,探究一个生物体内究竟有哪些基因、这些基因又如何通过“剪接”产生功能各异的转录本,是理解生命活动的核心,包括差异表达、可变剪接、RNA 变异鉴定等,而转录组组装,就是实现这一目标的关键一步。
转录组组装分为两种
参考基因组指导组装:准确度高,但必须依赖高质量参考基因组。
从头组装:完全不需要参考基因组,是非模式生物、癌症样本、稀有物种的重要解决方案。
传统短读长组装工具(如Trinity)虽成熟,但读段短、碎片化严重,无法还原全长转录本结构,真实剪接与异构体信息大量丢失。
随着三代长读长测序(ONT / PacBio)快速普及,全长转录本直接测序成为现实,能完整呈现异构体、基因融合、RNA 修饰等关键信息。但一个关键问题一直悬而未决:长读长从头组装工具越来越多(包括RATTLE、RNA-Bloom2、isONform),却始终缺乏系统、全面、贴近真实研究的基准测试,研究者无据可依。
为了填补这一空白,澳大利亚沃尔特和伊丽莎·霍尔医学研究所等团队在Genome Biology发表题为“A comprehensive evaluation of long-read de novo transcriptome assembly”的研究论文,该研究开展了迄今为止最全面、最贴近真实研究的长读长从头转录组组装基准测试。通过模拟数据、标准品掺入样本、多种真实生物样本(包括人类细胞系、单细胞数据和豌豆)的严格测试,本研究为缺乏高质量参考基因组的物种,提供了一份如何构建最优分析流程、从而进行可靠的差异表达分析的实用指南。

1. 研究设计
为评估长读长从头转录组组装工具的性能:该研究选取长读长从头转录组组装工具 isONform、RATTLE、RNA-Bloom2,以短读长标杆工具 Trinity 作对照,同时使用参考基因组指导工具 Bambu 进行组装与定量,并以其结果作为无参基因组数据集的评估参照。
对从头组装转录组的下游差异表达分析:采用Corset将转录本聚类为基因簇,通过minimap2将读段比对回组装转录组,使用Salmon进行转录本丰度定量(单细胞数据使用Oarfish),利用limma开展转录本和基因水平的差异表达分析。
五组独立数据集:覆盖多种测序技术、测序深度和物种,包括ONT cDNA模拟数据、人类肺癌细胞系ONT PCR-cDNA 数据(含标准品sequin spike-ins)、SG-NEx 项目的ONT直接 RNA 测序数据、PacBio Kinnex 单细胞RNA-seq数据、豌豆ONT PCR-cDNA数据。除单细胞数据外,所有数据集均配备匹配的短读长数据,利用 Trinity 进行分析。
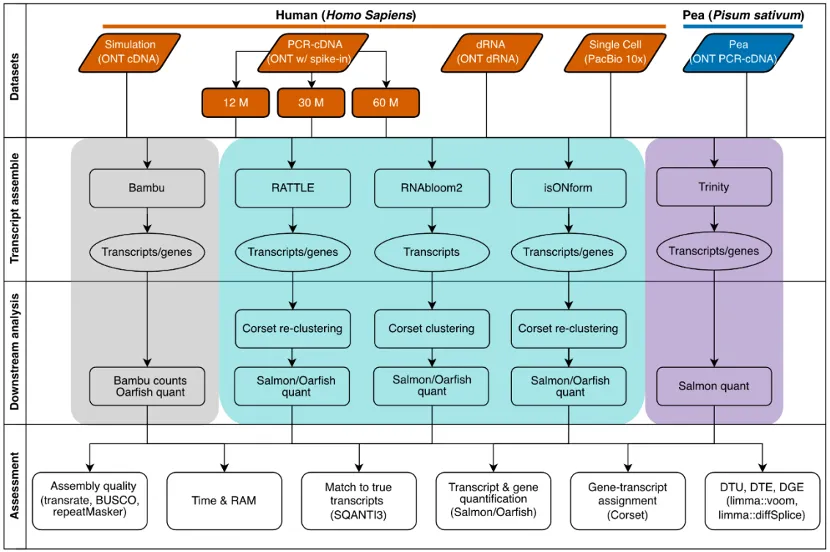
表1 基准测试所用数据集
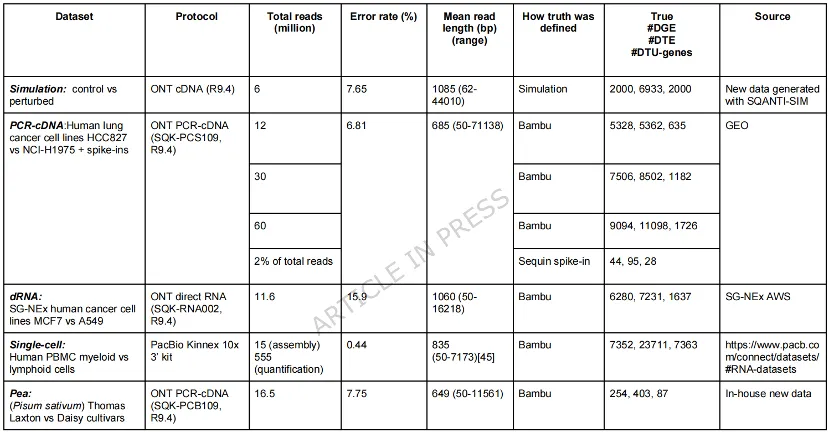
2. 计算效率
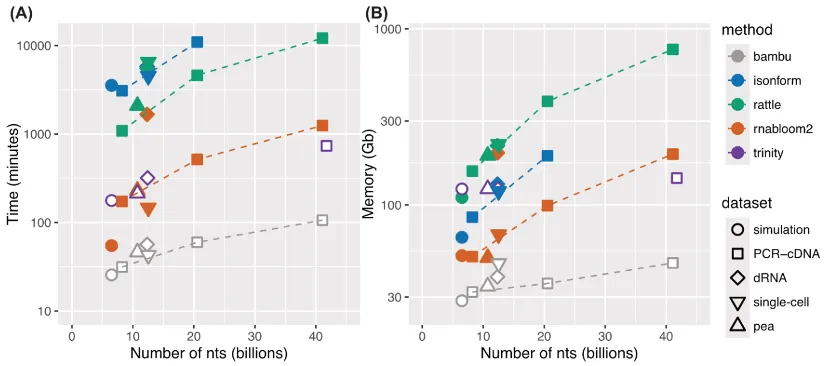
3. 组装质量
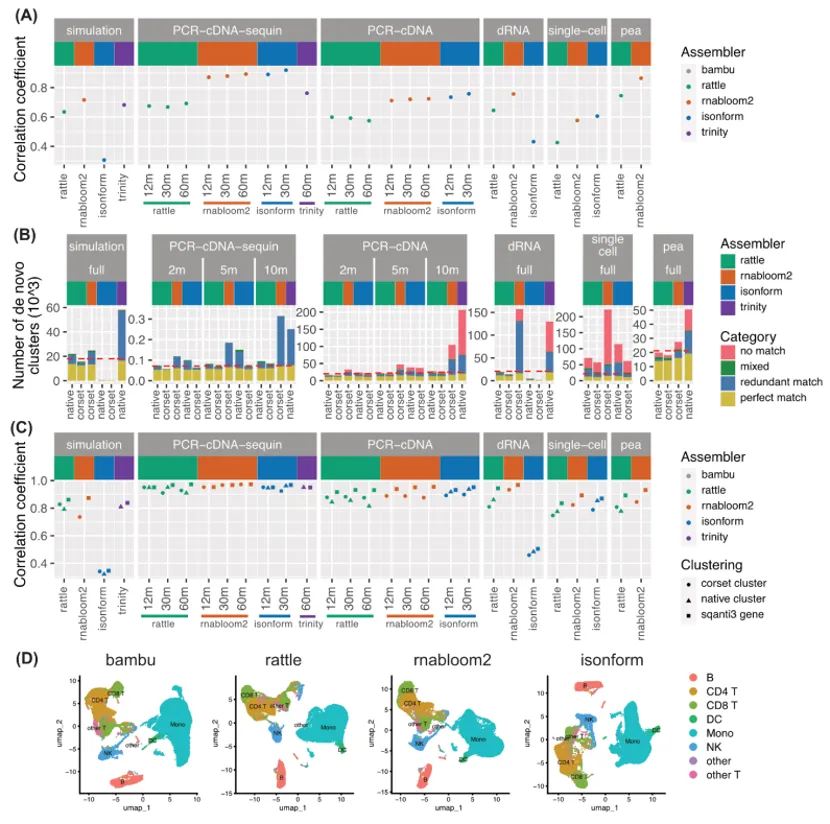
使用 SQANTI3、CRBB、BUSCO 等方法,结合参考基因组指导工具 Bambu 作为上限参照,短读长组装工具Trinity 作为对比,从转录本数量、长度、完整性、召回率等方面,评估各组装工具的转录本组装质量。
转录本数量差异极大:在模拟数据中RNA-Bloom2、RATTLE组装得到的转录本数(约40000条)和基因数(约17000个)相近,且与模拟设定的真实值(40509条转录本、18145个基因)接近;而isONform仅组装出4849条转录本和444个基因。在实测数据中RNA-Bloom2转录本最多但冗余度高,RATTLE数量最少,Trinity转录本数量最多,但以碎片化为主。测序深度提升长读长组装会增加新基因/新序列,但会降低平均转录本长度。
长读长在全长转录本上优势明显:Trinity平均长度最短、完整转录本最少,体现长读长在重构全长转录本上优于短读长。
召回率与保守性:RNA-Bloom2真实序列召回率更高,RATTLE更保守、遗漏部分转录本,提升深度仅能改善低表达基因恢复,二者在基因水平召回率相近。
假阳性与错误分析:模拟与PCR-cDNA标准品数据显示存在明显假阳性,此外,ONT数据存在插入缺失错误,导致BUSCO完整性下降,直接RNA数据尤为严重。PacBio与短读长无此问题,提示无参基因组分析需重视测序错误率。
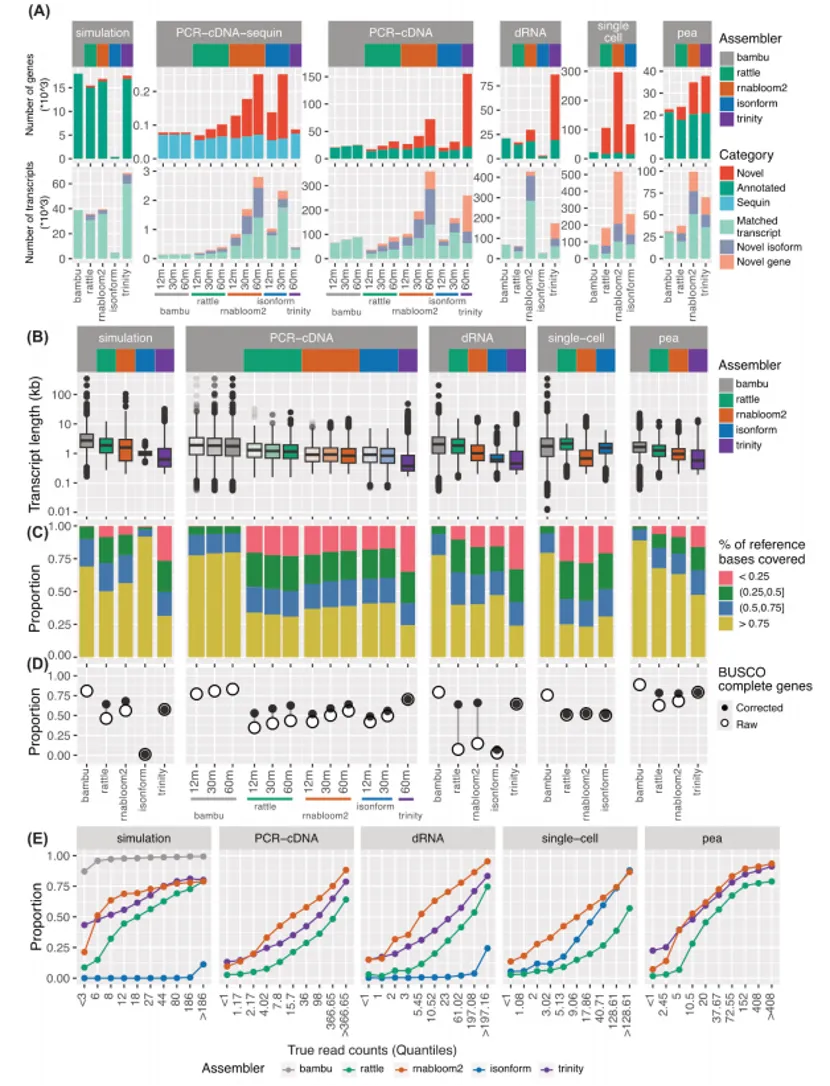
4. 转录本和基因丰度估计与差异表达分析
用 minimap2 比对、Salmon/Oarfish 定量,将组装转录本与真实表达量(模拟、标准品、Bambu 结果)对比,评估转录本与基因水平表达估计准确性。
转录本水平表达定量:RNA-Bloom2转录本计数与真实值的相关性始终位居前列;isONform在部分数据集表现最优,但前提是仅在运行成功时。Trinity在模拟数据与RNA-Bloom2相当,但在标准品数据中稍差。
基因水平聚类与定量:RNA-Bloom2和Trinity生成的基因簇最多,冗余性高(多个簇匹配同一参考基因)。多数从头组装工具基因水平表达相关性相近,RNA-Bloom2+Corset整体表现与RATTLE相当或更优。Corset聚类效果可靠,可替代原生聚类用于RNA-Bloom2下游分析。此外,增加测序深度对表达定量相关性提升很小。
单细胞分析:所有长读长组装工具均能重现参考基因组指导的细胞聚类模式,说明无参基因组也可实现准确的单细胞转录组分群,为缺乏参考基因组的非模式生物,实现单细胞分辨率的转录组分析提供了可能。
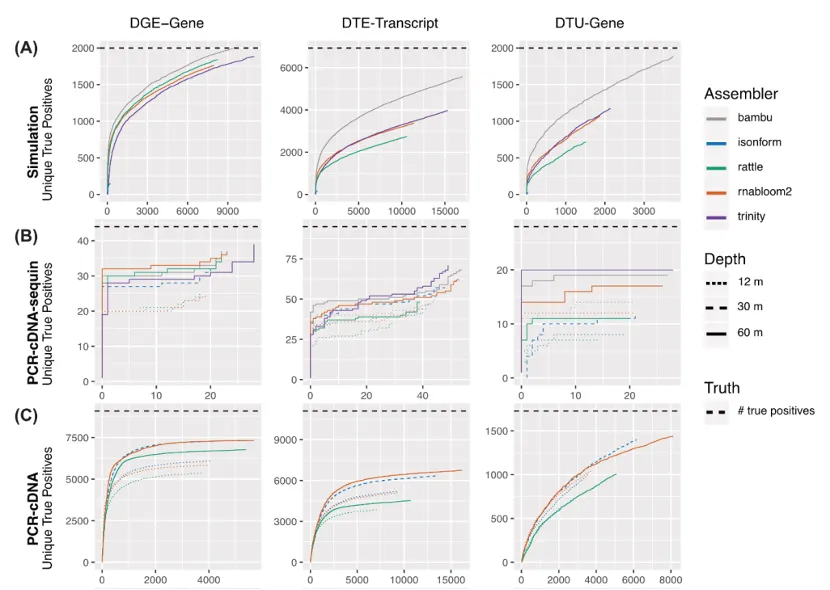
基于前面得到的计数结果,使用limma开展差异表达分析,重点评估各工具在差异表达基因(DGE)、差异表达转录本(DTE)、转录本使用差异(DTU)上的识别能力。
差异表达基因(DGE):
在模拟数据中所有方法表现良好,接近Bambu,Trinity略逊于长读长工具。
在标准品数据中Trinity和RNA-Bloom2略优于其他工具。
在实测数据集中RNA-Bloom2表现始终位居前列,isONform在成功运行的数据集上性能与RNA-Bloom2 相当甚至更优。
差异表达转录本(DTE):
长读长工具中,RNA-Bloom2在各数据集上表现最优,Trinity在模拟数据和标准品数据中表现同样良好。
转录本使用差异(DTU):多数数据集下RNA-Bloom2仍是最优的从头组装工具。
为探究混合组装方法是否能整合长、短读长测序的优势,表现优于单一测序技术。研究选用两种主流混合工具:
RNA-Bloom2混合模式:先利用短读长数据对长读长数据进行抛光(纠错),再进行组装。
rnaSPAdes:先利用短读长数据构建组装图谱,再将长读长数据比对至图谱以解析重复序列并填补缺口。
本研究全面评估了三款长读长从头转录组组装工具(isONform、RATTLE、RNA-Bloom2),以Trinity和 Bambu为对照,在多类型数据集上验证了无参考基因组差异分析的潜力与问题。
计算性能仍是主要瓶颈,多数工具耗时长、占内存大,难以应对未来超大数据集。RNA-Bloom2整体最优,搭配Corset聚类适合下游差异分析,但转录本冗余度高。RATTLE结果保守、计算需求高。isONform小数据集表现好,但对数据鲁棒性差、运行极慢。长读长可获得更完整的全长转录本,但ONT测序错误高,影响下游分析。短读长碱基质量高、差异检测统计效力强,但转录本碎片化,异构体解析能力弱。