 夜雨聆风
夜雨聆风





AI 让 H&E 病理切片“预测”空间蛋白质图谱:肺癌可解释生物标志物发现新路径
文章标题
AI-enabled virtual spatial proteomics from histopathology for interpretable biomarker discovery in lung cancer。 (Nature)
杂志
Nature Medicine,Volume 32,Pages 231–244,2026。(Nature)
发表时间
在线发表时间:2026 年 1 月 5 日;正式版本日期:2026 年 1 月 5 日;期刊卷期时间:2026 年 1 月。(Nature)
摘要
空间蛋白质组学能够以高分辨率绘制蛋白表达分布,并有望改变人们对生物学和疾病的理解,但其临床转化仍受到成本、复杂性和可扩展性的限制。本文提出 H&E to protein expression(HEX)模型,这是一种可从常规 H&E 病理切片中计算生成空间蛋白质组图谱的人工智能模型。HEX 在来自 382 个肿瘤样本、匹配蛋白表达信息的 819,000 个组织病理图像块上训练和验证,能够准确预测覆盖免疫、结构和功能程序的 40 种生物标志物表达。研究者进一步建立了整合原始 H&E 图像与 AI 推断虚拟空间蛋白质组的多模态方法,用于提升结局预测能力。在 6 个独立非小细胞肺癌队列、共 2,298 名患者中,HEX 驱动的多模态整合相较传统临床病理和分子标志物,使预后预测准确性提升 22%,免疫治疗反应预测提升 24–39%。生物学解释显示,治疗反应相关的肿瘤—免疫空间生态位具有明确组织结构:应答者中可见辅助性 T 细胞与细胞毒性 T 细胞共定位,非应答者中则可见免疫抑制性肿瘤相关巨噬细胞和中性粒细胞聚集。总体而言,HEX 为研究空间生物学提供了一种低成本、可扩展的方法,并有助于发现和转化精准医学中的可解释生物标志物。(Nature)
引言
空间生物学正在成为基础研究和精准医学的重要方向。传统空间蛋白质组技术可以在组织中同时检测多个蛋白标志物,并保留细胞与组织结构的空间信息,但这类技术通常依赖昂贵设备、复杂实验流程和专业人员,因此难以在大规模临床样本中普及。与之相比,H&E 染色病理切片是临床诊断中最常见、最标准化的材料,几乎所有癌症患者的诊疗流程都会产生这类图像。近年来,深度学习已经能够从 H&E 图像中推断突变、基因表达等分子特征,但既往模型多以整块组织或整张切片作为训练标签,难以捕捉肿瘤内部高度异质的空间分布。
本文的核心问题是:能否仅用常规 H&E 病理图像,推断出高维空间蛋白表达图谱,并进一步用于肺癌预后和免疫治疗疗效预测?作者提出 HEX 模型,利用病理基础模型从 H&E 图像中预测 40 种蛋白生物标志物的空间表达,并通过 MICA 多模态整合框架,将原始组织形态与 AI 生成的虚拟 CODEX 空间蛋白质组信息结合起来。研究不仅验证了模型对蛋白表达预测的准确性,也在多个独立非小细胞肺癌队列和泛癌种队列中检验其临床预测价值,进一步尝试解释哪些肿瘤—免疫空间结构与患者结局和免疫治疗反应相关。(Nature)
研究内容
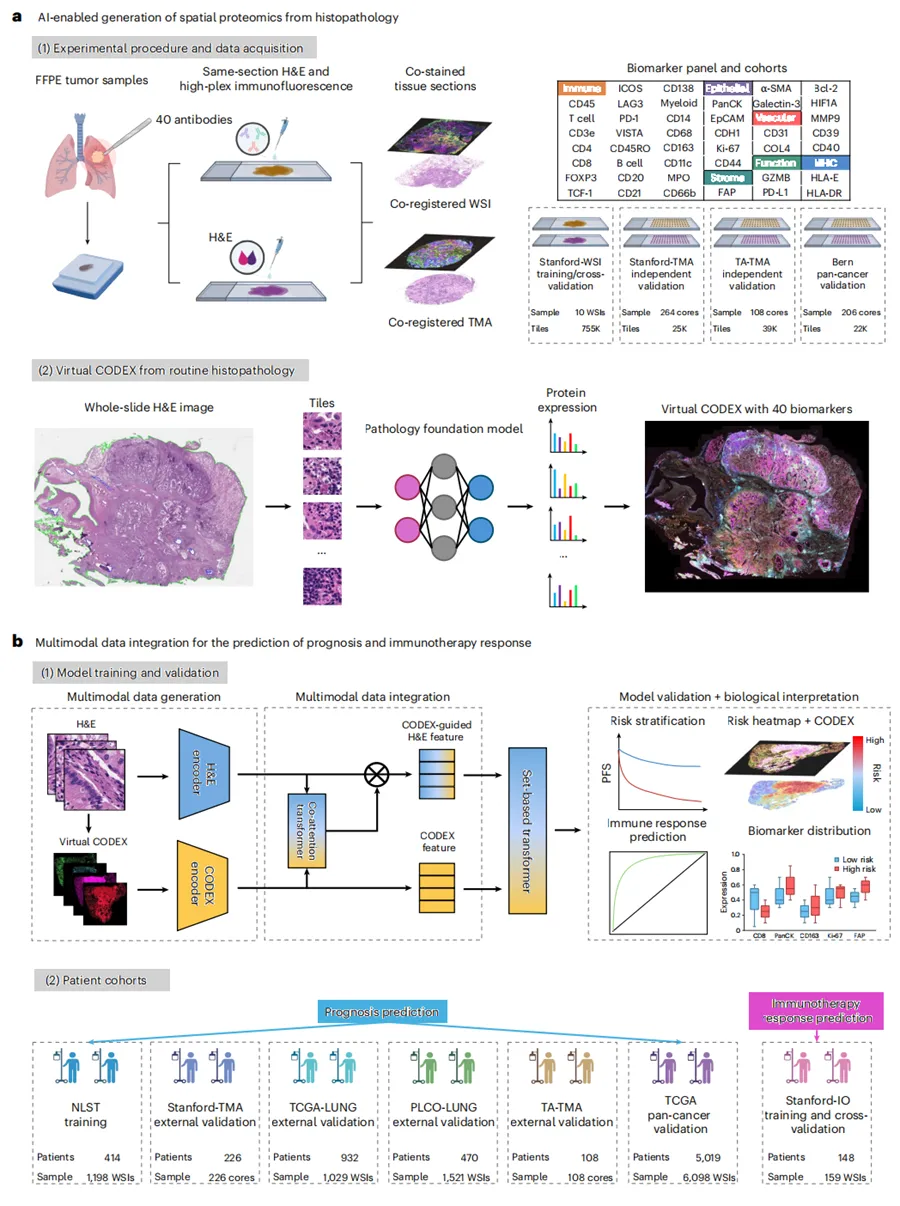
Fig. 1|HEX 的开发、验证与临床应用。a,HEX 的开发和技术验证。HEX 是一种预测型 AI 模型,旨在从标准 H&E 染色切片生成高维空间蛋白质组图谱。HEX 使用来自 10 名 NSCLC 患者肿瘤样本的数据进行训练;这些样本在同一组织切片上进行 H&E 染色和采用 40 抗体面板的高通量免疫荧光染色,并由此获得超过 755,000 个具有匹配蛋白表达信息的图像块。HEX 利用先进的病理基础模型,根据 H&E 图像同时预测 40 种蛋白生物标志物表达,从而由标准组织病理图像生成虚拟空间蛋白质组图谱。蛋白表达预测准确性在两个独立数据集中验证,这两个数据集包含 372 个具有共染组织切片的肿瘤样本。为评估泛化能力,HEX 还在一个泛癌种数据集上进行外部验证,该数据集包含来自 34 种组织类型的 206 个肿瘤样本,具有 57-plex CODEX 图像和匹配 H&E 切片,且采用不同染色流程和扫描仪成像。
b,临床验证和生物学解释。HEX 的临床相关性在 5 个 NSCLC 队列、共 2,150 名患者中用于预后预测,并在 TCGA 中 12 个其他癌种、共 5,019 名患者中进一步评估。HEX 还在另一个接受免疫检查点抑制剂治疗的 148 名患者队列中用于治疗反应预测。研究者开发了一种多模态数据整合方法,将原始 H&E 图像和 AI 推断的虚拟空间蛋白质组图谱结合起来,以进一步增强结局预测。HEX 生成的生物标志物分布揭示了肿瘤—免疫生态位的空间模式,为临床结局预测提供生物学依据和解释。FFPE,福尔马林固定石蜡包埋。示意图元素使用 BioRender.com 创建。(Nature)
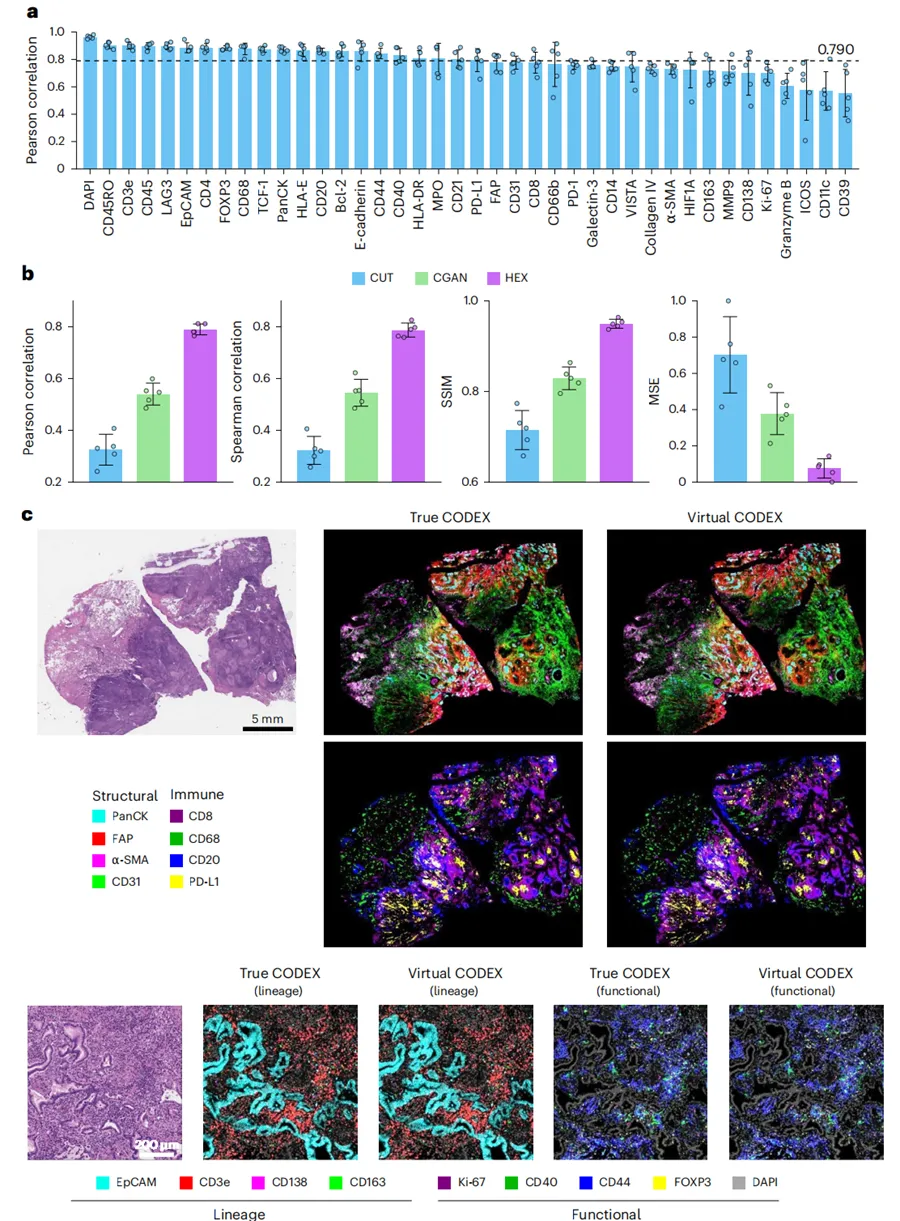
Fig. 2|HEX 用于蛋白生物标志物预测的性能评估。a,HEX 在使用 10 张全切片图像进行交叉验证时,对 40 种蛋白生物标志物的预测性能。结果显示,实测与预测的生物标志物分布具有高度一致性。b,HEX 与两种生成式 AI 模型在多种性能指标上的比较。c,HEX 预测的虚拟空间蛋白质组图谱代表性可视化。虚拟 CODEX 图像在结构、免疫、谱系和功能生物标志物方面均与真实图像高度相似,并且在标准空间分辨率和精细空间分辨率下均如此,对应的图像块大小分别为 224 像素和 14 像素。类似结果在 10 张独立全切片图像中获得。在 a 和 b 中,柱形表示 Stanford-WSI 数据集中五折交叉验证的均值(n = 10 张 WSI),数据点表示各个折,误差线表示标准差。(Nature)
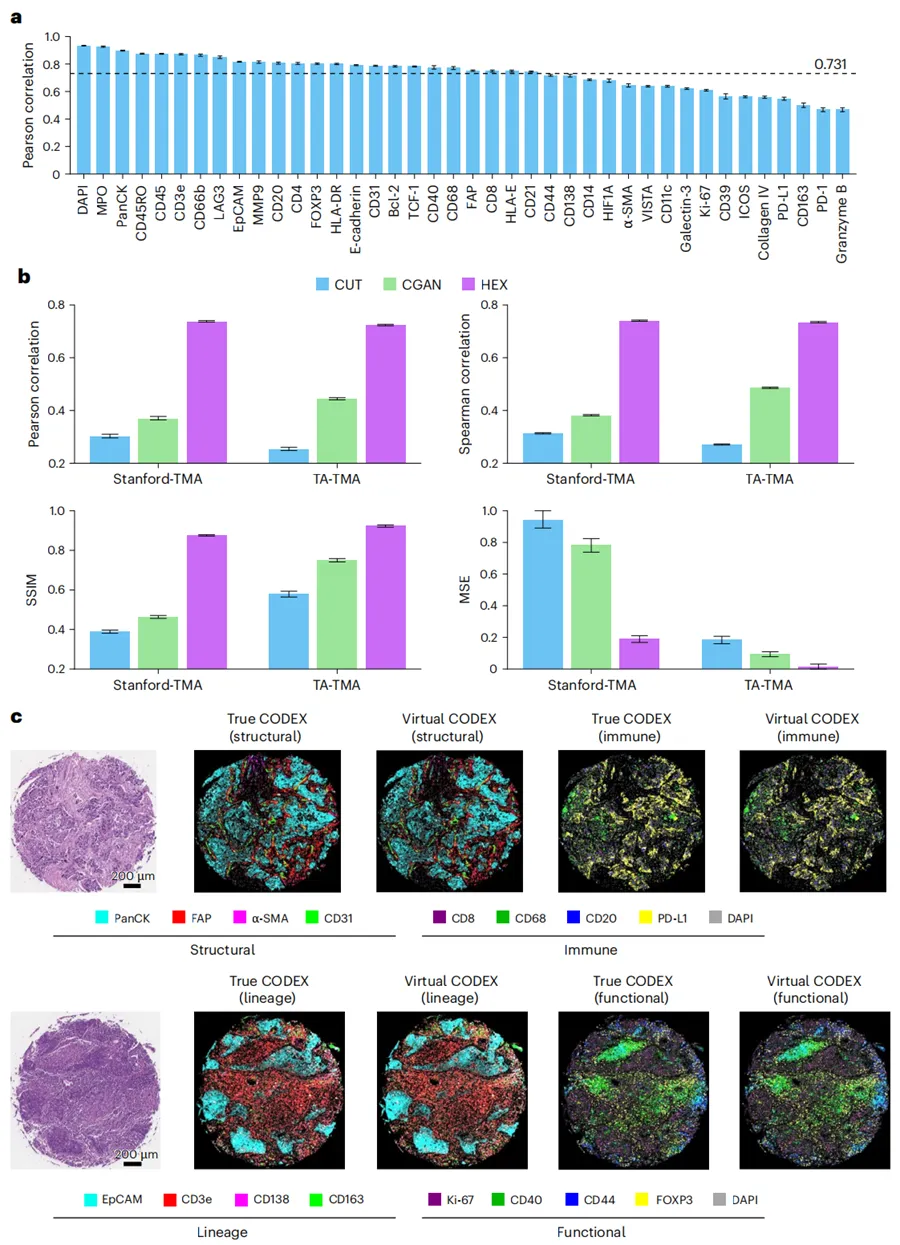
Fig. 3|HEX 用于蛋白生物标志物预测的独立验证。a,HEX 在包含 372 个 TMA 核心的独立测试集上预测 40 种蛋白生物标志物的性能,其中 Stanford-TMA 为 n = 264,TA-TMA 为 n = 108。b,HEX 与两种生成式 AI 模型在多种性能指标上的比较。c,HEX 预测的虚拟空间蛋白质组图谱代表性可视化。虚拟 CODEX 图像在结构、免疫、谱系和功能生物标志物方面均与真实图像高度相似。可视化使用 14 像素图像块大小的高分辨率 HEX 版本生成,以增强空间细节。类似结果在 372 个核心中获得。在 a 和 b 中,柱形表示点估计值,误差线表示 95% bootstrap 置信区间(n = 1,000 次重采样)。(Nature)
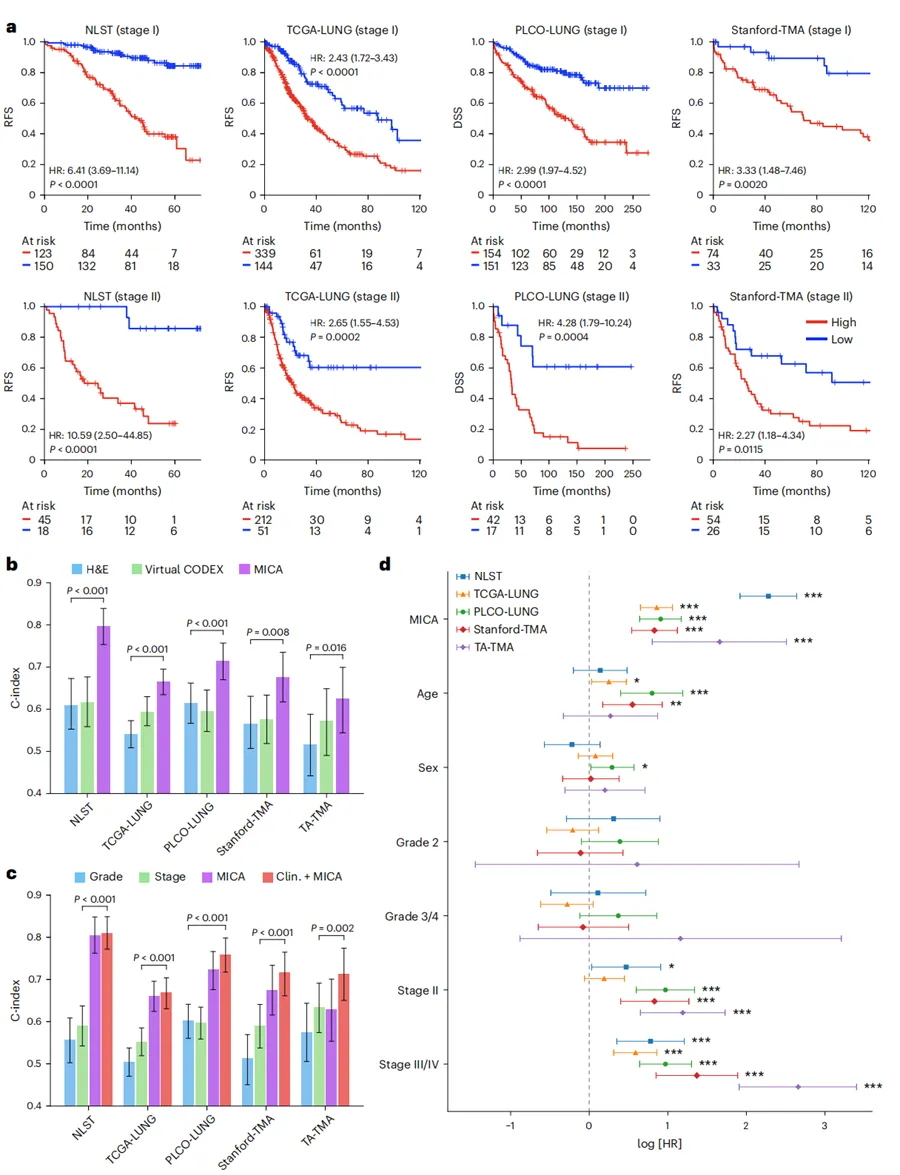
Fig. 4|MICA 改善早期 NSCLC 患者的预后预测。a,MICA 预测风险分组在 I 期和 II 期 NSCLC 患者中的 Kaplan–Meier 分析。患者在 4 个独立队列中被显著分为高风险组和低风险组,分别以红色和蓝色表示,结局指标为 RFS 或 DSS。所有验证队列使用相同截断值,该截断值基于 NLST 训练队列确定。每组在各时间点的风险人数显示在每张图下方。统计显著性采用双侧 log-rank 检验确定。b,早期 NSCLC 五个队列中预后模型的性能比较。c,早期 NSCLC 中 MICA 风险预测与临床特征(Clin.)的性能比较。在 b 和 c 中,柱形表示点估计值,误差线表示 95% bootstrap 置信区间(n = 1,000 次重采样)。d,在五个队列中纳入临床协变量和 MICA 预测风险评分的多变量 Cox 回归分析,这些队列包括 NLST(n = 336 名患者)、TCGA-LUNG(n = 746)、PLCO-LUNG(n = 364)、Stanford-TMA(n = 187)和 TA-TMA(n = 94)。点表示 log[HR] 点估计值,水平线表示 95% 置信区间。MICA 风险评分仍然是 RFS、DSS 或总生存的显著独立预测因子。统计显著性采用双侧 Wald 检验确定,P 值未经校正。***P < 0.001;**P < 0.01;*P < 0.05。(Nature)
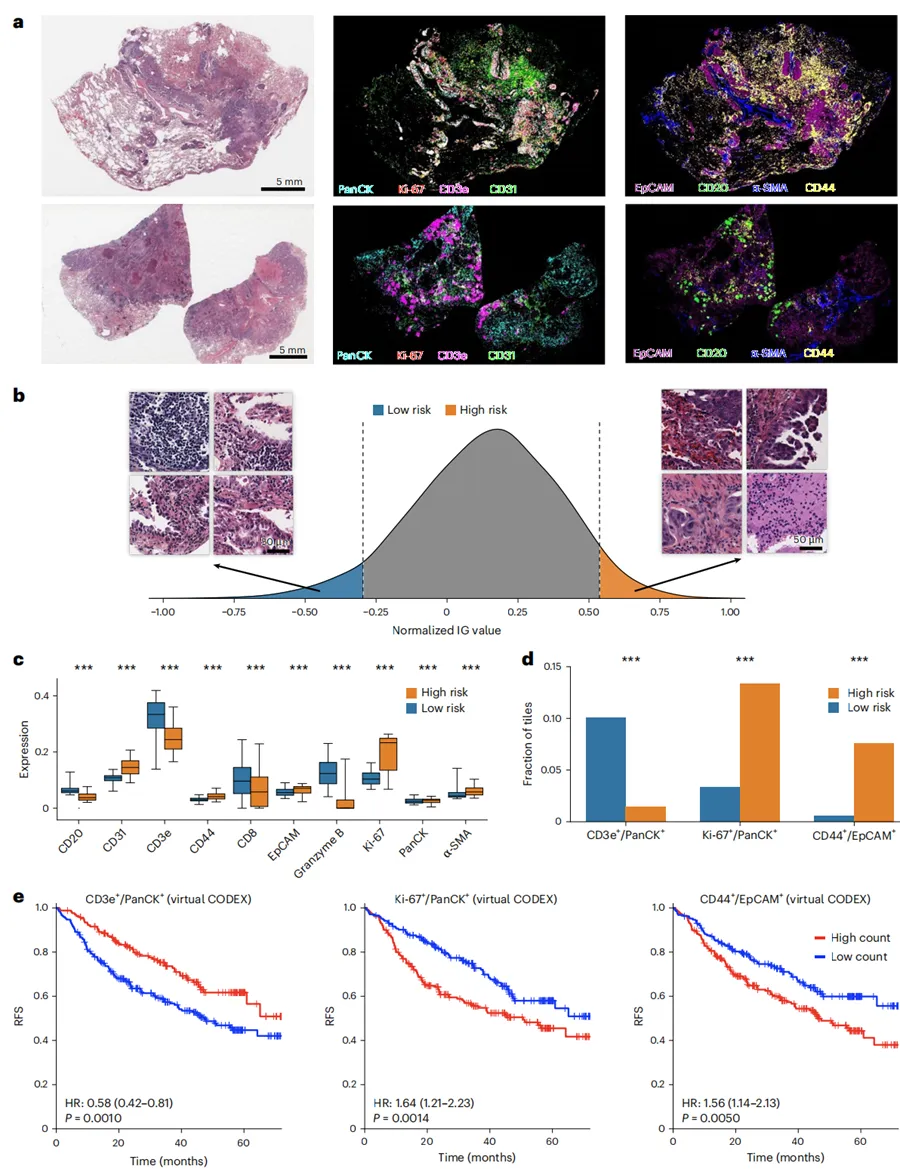
Fig. 5|使用 HEX 虚拟空间蛋白质组解释 MICA 推导的风险预测。a,NLST 队列中由 MICA 模型预测的一名高风险患者(上)和一名低风险患者(下)的代表性示例。对于每名患者,显示原始 H&E 图像以及 HEX 生成的部分生物标志物对应虚拟 CODEX 图。b,所有图像块总体 integrated gradient(IG)值的直方图。Integrated gradient 值对应每个图像块的预测风险评分。高风险和低风险组由较高和较低 integrated gradient 值定义。c,箱线图比较高风险组和低风险组中所选蛋白生物标志物的表达分布,高风险组 n = 26,519 个图像块,低风险组 n = 26,416 个图像块。中心线表示中位数,箱体表示四分位距(第 25–75 百分位),须延伸至第 5–95 百分位。组间差异采用双侧 Mann–Whitney U 检验评估。d,比较高风险组和低风险组中三对生物标志物(CD3e+/PanCK+、Ki-67+/PanCK+ 和 CD44+/EpCAM+)高共表达值图像块的比例。采用双侧卡方检验评估组间统计差异。***P < 0.001。e,基于 c 中所选生物标志物对高共表达模式图像块数量的Kaplan–Meier 分析。患者被分为高计数组和低计数组。统计显著性采用双侧 log-rank 检验计算,未进行多重比较校正。(Nature)
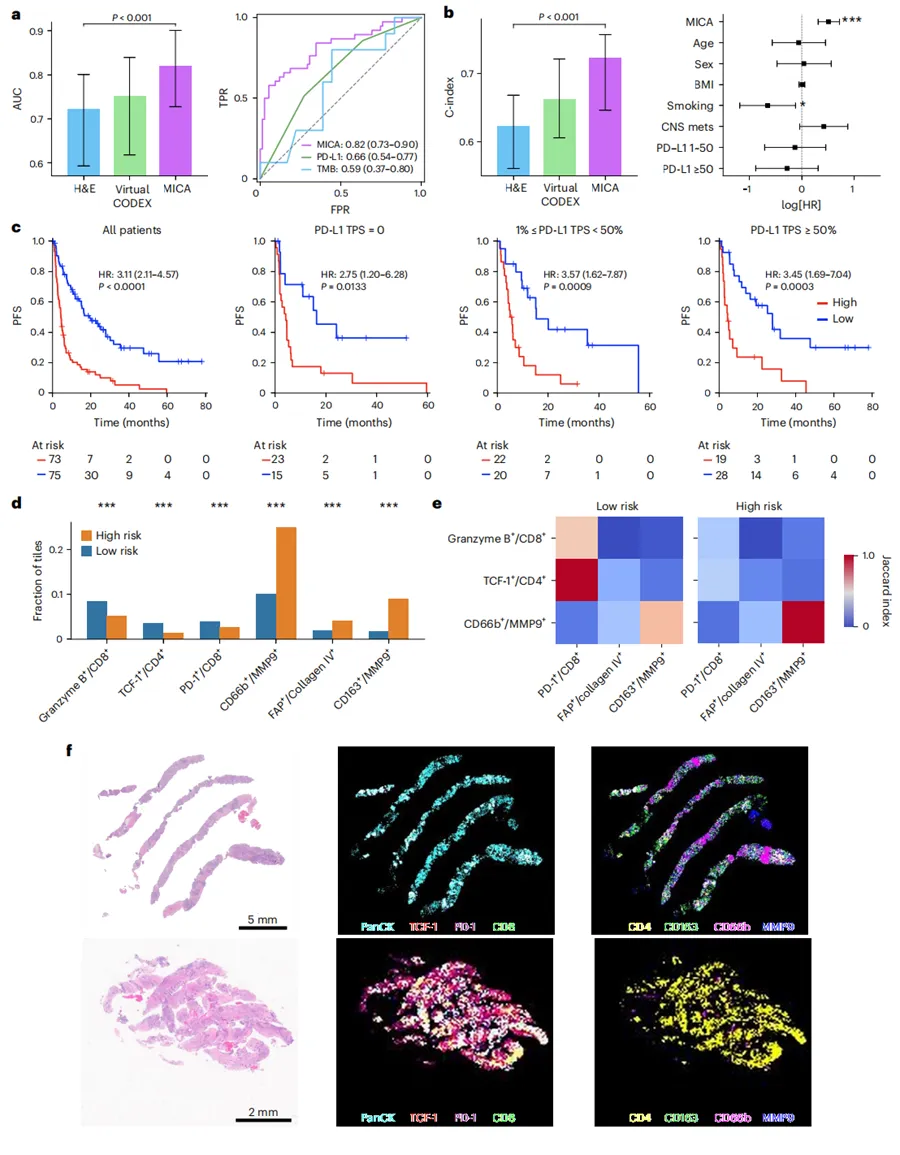
Fig. 6|MICA 改善晚期 NSCLC 的免疫治疗反应预测,并识别空间蛋白质组特征。a,预测客观反应的模型性能比较。左侧显示基于 H&E、虚拟 CODEX 和 MICA 的模型 AUC 点估计值。右侧显示 MICA 与 PD-L1 表达和 TMB 比较的 ROC 曲线。b,预测 PFS 的模型性能比较。左侧显示基于 H&E、虚拟 CODEX 和 MICA 的模型 C-index 点估计值。右侧为森林图,显示包含 MICA 推导风险评分和临床协变量的多变量 Cox 回归。点表示 log[HR] 点估计值,水平线表示 95% 置信区间。统计显著性采用双侧 Wald 检验确定。a 和 b 中柱状图的误差线表示由聚合患者水平风险预测得到的 95% bootstrap 置信区间(n = 1,000 次重采样),统计显著性采用双侧Wilcoxon 符号秩检验评估。c,按 MICA 预测风险评分在所有患者及 PD-L1 表达亚组中分层的 PFS Kaplan–Meier 分析,PD-L1 亚组包括肿瘤比例评分(TPS)= 0、1% ≤ TPS < 50% 和 TPS ≥ 50%。每组在各时间点的风险人数显示在每张图下方。高风险组和低风险组之间的生存差异采用双侧 log-rank 检验评估(截断值:中位数)。d,高风险组和低风险组中,所选生物标志物对定义的免疫和基质细胞状态图像块比例,包括 granzyme B+/CD8+、TCF-1+/CD4+、CD66b+/MMP9+、PD-1+/CD8+、FAP+/collagen IV+ 和 CD163+/MMP9+。采用双侧卡方检验评估统计差异(***P < 0.001)。e,SCS 的标准化 Jaccard 指数。该分析揭示了高风险组和低风险组中关键细胞类型不同的空间共定位模式。f,MICA 模型预测的一名高风险患者(上)和一名低风险患者(下)的代表性示例。对于每名患者,显示原始 H&E 图像以及 HEX 生成的所选生物标志物对应虚拟 CODEX 图。BMI,体重指数;CNS mets,中枢神经系统转移;FPR,假阳性率;TPR,真阳性率。(Nature)
结论
从读者角度看,这篇论文的核心结论是:常规 H&E 病理切片中包含足以推断空间蛋白表达的信息,AI 可以把这些形态学信息转化为近似空间蛋白质组的“虚拟 CODEX”图谱。HEX 不只是一个图像生成或标志物预测模型,它还通过 MICA 框架与原始病理图像联合使用,在非小细胞肺癌中提升了预后预测和免疫治疗反应预测能力。更重要的是,模型给出的预测可以被空间蛋白表达模式解释,例如低风险或免疫治疗应答相关样本中更常见免疫细胞浸润和 T 细胞相关共定位,而高风险或非应答样本中更常见增殖性、基质性或免疫抑制性空间生态位。这提示基于常规病理切片的虚拟空间蛋白质组可能成为连接临床病理工作流和空间生物学发现的一种可扩展路径。
全文总结
本文提出 HEX 模型,用标准 H&E 病理图像预测 40 种蛋白标志物的空间表达,并生成虚拟 CODEX 空间蛋白质组图谱。研究首先在同一组织切片的 H&E 与 CODEX 共染数据上训练模型,再通过独立 TMA 队列和 Bern 泛癌种数据集验证其泛化能力。结果显示,HEX 在蛋白表达预测上优于既有生成式模型,并能在不同组织类型、不同染色流程和不同成像平台上保持较强性能。随后,作者提出 MICA 多模态整合框架,将 H&E 形态特征与 HEX 生成的虚拟空间蛋白质组整合,用于临床结局预测。在早期 NSCLC 中,该方法相较单独 H&E、单独虚拟 CODEX 或传统临床病理因素,表现出更好的预后预测能力;在接受免疫检查点抑制剂治疗的晚期 NSCLC 中,也优于 PD-L1 和 TMB 等常用标志物。生物学解释进一步显示,模型识别的风险和治疗反应并非黑箱结果,而是与特定肿瘤—免疫空间结构相关。总体而言,这项研究将常规病理图像、空间蛋白质组学和可解释 AI 结合起来,为低成本、大规模空间生物标志物发现及临床转化提供了新方案。