 夜雨聆风
夜雨聆风





AI正在重写单细胞与空间转录组吗?一篇2026综述讲透了机会与边界
这两年,AI 在生物信息学里的存在感越来越强。尤其是在单细胞测序和空间转录组领域,几乎每隔一段时间就会冒出一批新模型:scGPT、Geneformer、scVI、GraphST、MultiVI……名字越来越多,架构越来越复杂,宣传也越来越猛。
但真正的问题其实只有一个:
AI 到底是在帮我们更接近真实生物学,还是只是把分析流程变得更“高级”了?
2026 年发表的一篇综述《Applications of AI to single-cell and spatial transcriptomics: current state-of-the-art and challenges》https://doi.org/10.3389/fbinf.2025.1715821做了一件很重要的事:它没有停留在“模型很新”“结构很强”这种技术兴奋里,而是回到研究者最关心的场景——在单细胞和空间转录组的真实分析任务里,AI 到底值不值得用,哪里该用,哪里不该迷信。
一、为什么单细胞和空间转录组,成了AI最爱“扎堆”的地方?
原因并不神秘:这类数据天然就“又大又复杂”。
单细胞 RNA-seq 往往动辄成千上万到几十万细胞,每个细胞又有上万基因表达;空间转录组在此基础上还额外叠加了空间坐标、组织图像,甚至免疫荧光等多模态信息。也就是说,研究者面对的不是单一矩阵,而是一个同时包含表达、位置、形态和上下文的复杂系统。
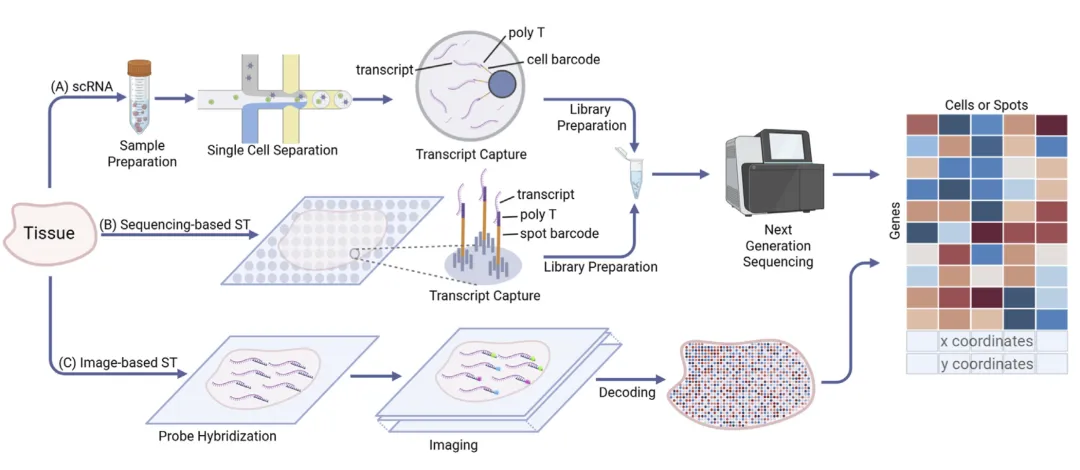
从组织样本出发,scRNA-seq、测序型空间转录组和成像型空间转录组分别通过不同路径生成表达数据;其中空间转录组额外保留了空间位置信息,这也是后续AI模型能够整合表达、空间与图像信息的基础。
这正是深度学习最擅长的地方:它不怕高维,不怕异构,也擅长把不同类型的数据压到同一个表示空间里。因此,这篇综述并不否认 AI 的重要性,反而承认它在多模态整合、图像融合、空间建模方面确实有天然优势。问题不在于 AI 能不能做,而在于:它做出来的结果,能不能真的用于生物学发现。
二、AI到底是怎么“读”单细胞数据的?
很多人会误以为,AI 模型就是把基因表达矩阵直接塞进去,然后吐出一个结果。其实不是。
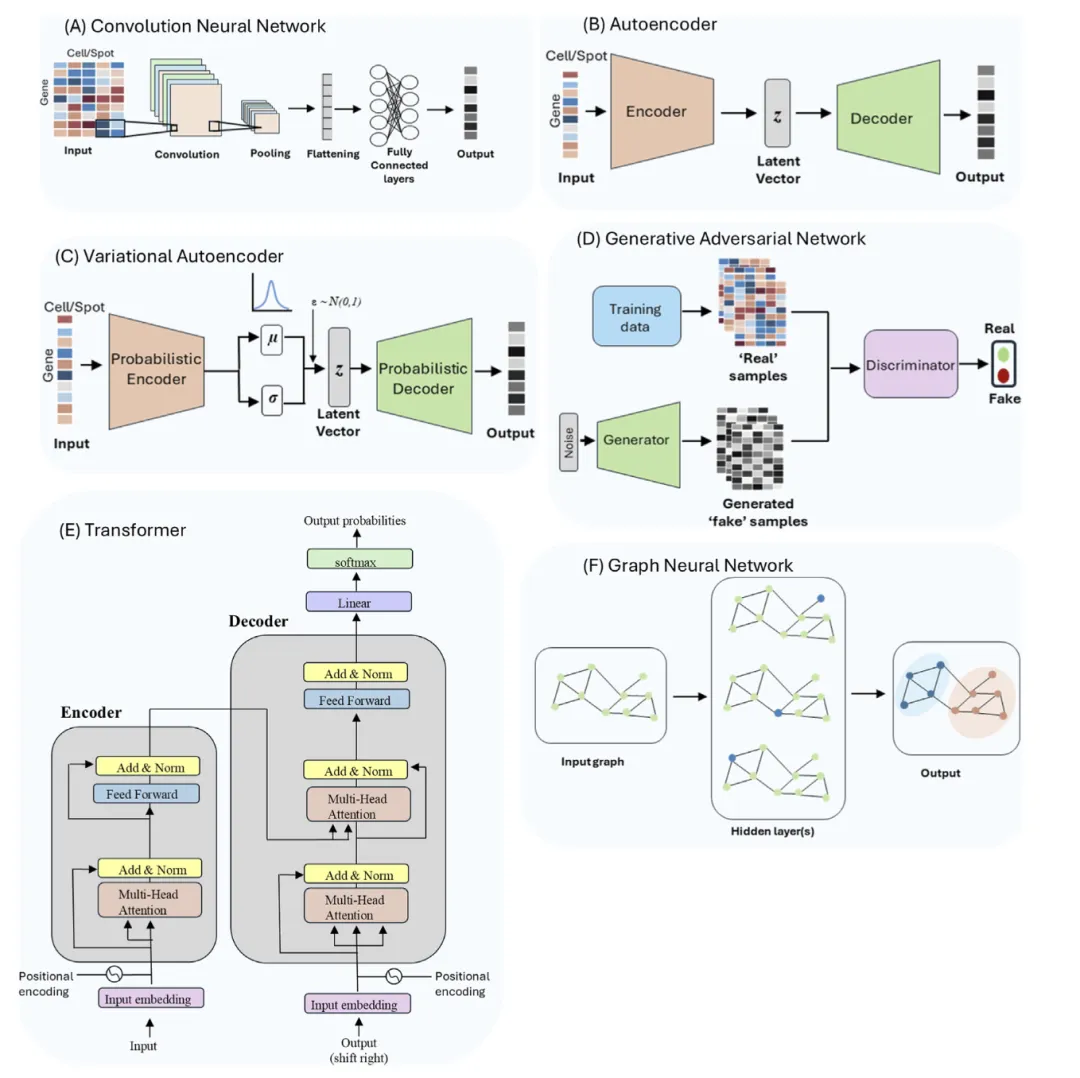
图2|单细胞与空间转录组分析中常见的深度学习架构。
包括卷积神经网络(CNN)、自编码器(AE)、变分自编码器(VAE)、生成对抗网络(GAN)、Transformer 和图神经网络(GNN)。不同模型各有分工:有的擅长降维与表示学习,有的适合生成与补全,有的更适合处理图像、空间邻接关系和多模态整合。
在这篇综述里,作者专门梳理了几类主流架构。最容易理解的一类是 Autoencoder / VAE:它们会把高维表达矩阵压缩到一个低维潜空间,再从这个潜空间重建原始数据。这样做的好处是,模型可以学习复杂的非线性结构,还能更自然地适配单细胞数据常见的稀疏性、过度离散和技术噪声。scVI 就是这一路线里最成功、也最被广泛使用的代表。
另一类是 Transformer 基础模型。它并不是像看自然语言那样“直接看基因”,而是先把基因表达转成模型能处理的 token。常见做法包括:按表达量排序,把基因当成有顺序的 token;把表达量分箱后离散化;或者直接把表达值投影成可学习的向量。不同方案各有代价:排序法更稳,但会损失定量信息;分箱法保留了大体尺度,却会牺牲细粒度差异;直接数值投影保留信息最完整,但建模更难。
再往空间转录组走,GNN 和 CNN就更自然了。因为空间转录组天生就有邻接关系、有图像信息,图神经网络可以直接建模细胞或 spot 之间的空间邻近,卷积网络则能提取 H&E 切片里的组织形态特征。也正因为如此,DL 在空间组学里往往比在普通 scRNA-seq 里更有“用武之地”。
三、哪些任务里,AI是真的有价值的?
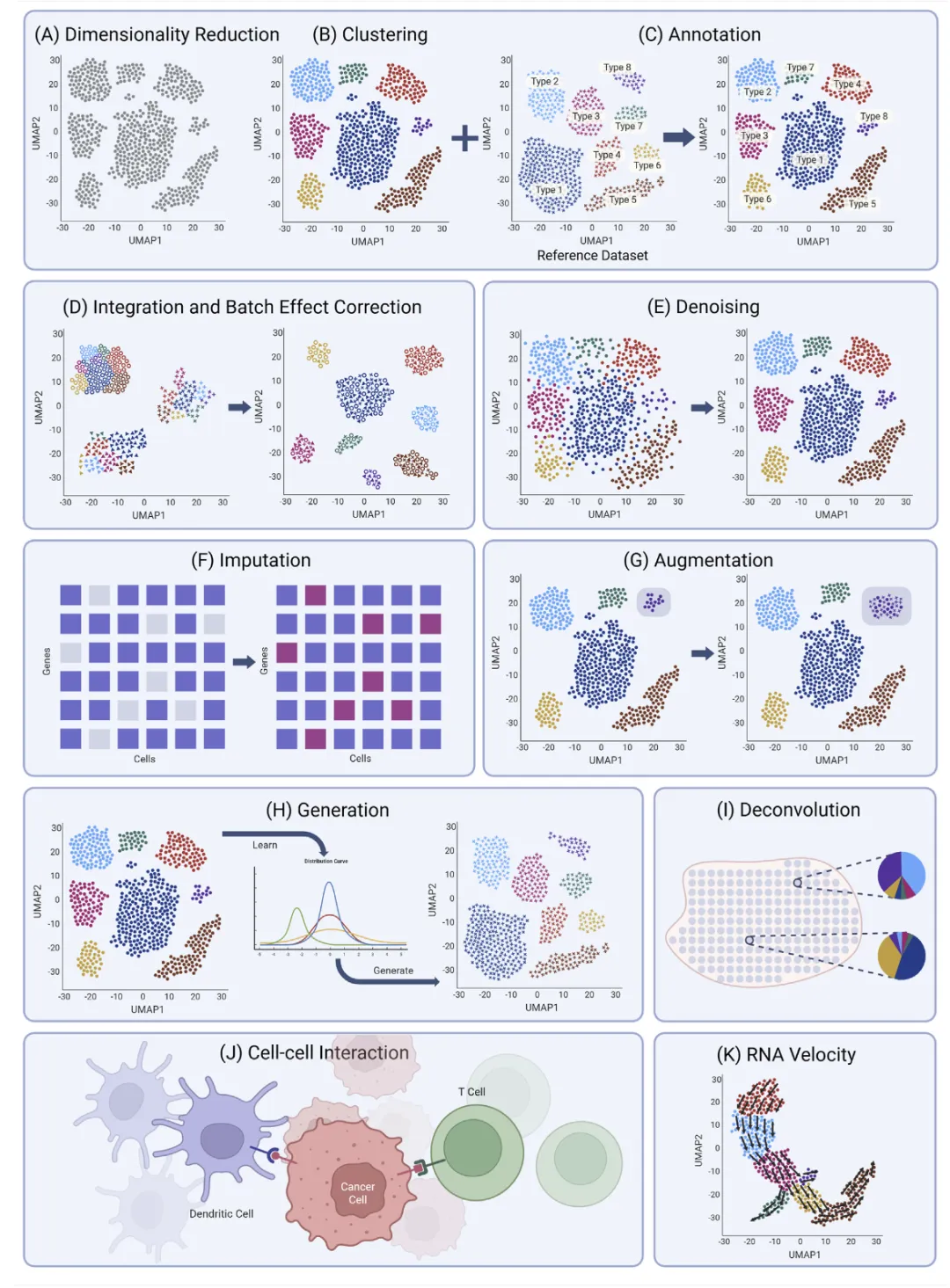
图3|深度学习在单细胞与空间转录组中的主要应用任务。
包括批次整合、去噪、插补、数据增强、数据生成、空间去卷积、细胞通讯分析和RNA velocity 等。需要注意的是,并不是所有任务都同样适合AI:有些场景中AI能显著提升多模态整合能力,但在差异分析等强调严格统计解释的任务中,过度依赖AI反而可能引入伪信号。
先说结论:不是所有任务都适合AI,但有几类任务,AI确实很强。
第一类是多组学整合。论文指出,在普通 scRNA-seq 批次整合里,DL 并没有稳定压过 Harmony 这类成熟方法;但在 scRNA + scATAC、RNA + protein、空间信息 + 表达信息这种跨模态整合任务里,DL 的表现更有说服力。原因很简单:不同模态统计分布差异太大,传统方法往往难以优雅地压到同一个空间,而 VAE、图模型和注意力机制更容易处理这件事。
第二类是空间转录组的区域识别和图像融合。这篇综述明确提到,如果你要把组织学图像、空间坐标和基因表达一起纳入分析,DL 几乎是绕不开的;其中,GraphST 被作者点名为当前表现最好的 DL 方法之一。也就是说,AI 在这里不是“锦上添花”,而更像是“结构上更合适”。
第三类是自动注释的第一轮筛查。像 scGPT、CellFM 这样的基础模型,可以把新数据快速投射到已有参考图谱里,先给出一个初步注释和置信度分数。在大规模人类数据里,这种“先粗分、再人工修正”的工作流是有现实意义的,尤其适合加快初筛。文中甚至建议:如果你并不清楚该选哪种方法,而又是做人类数据,可以优先考虑 scGPT,因为它的基准测试相对更充分。
四、哪些地方最容易被AI“带偏”?
这篇综述最有价值的地方,不是它夸 AI,而是它很清楚地指出了高风险区。
最典型的一类,就是去噪和插补。
DCA、scVI、DeepImpute 这些方法在某些任务上确实能把数据“变好看”,比如恢复被人为破坏的表达值、提高自动注释效果,甚至让某些 marker 看起来更清晰。但问题是,一旦把这些“修复后”的数据直接拿去做差异表达、基因相关性、marker 挖掘、细胞通讯分析,假阳性就会显著增加。作者的措辞其实很明确:在 discovery research 里,单细胞去噪仍然是一个有争议且较少被真正采用的方向。
第二个高风险区,是数据增强和生成式补样。如果用 cscGAN、scGFT 这类方法去扩充稀有细胞群,的确可能帮助聚类和轨迹分析更平滑;但本质上你是在人工放大样本信息量,这很可能会抬高统计检验的 power,从而推高 I 类错误。说得直白一点:它也许能让图更漂亮、轨迹更顺,但未必能让结论更真。
第三个问题是自动注释的泛化能力。很多模型在论文里准确率很高,但那往往是因为训练集和测试集来自同一个大数据集切分,真实世界里常见的批次差异、疾病状态差异并没有充分暴露出来。综述里提到,哪怕是表现不错的 scGPT,一旦 query 数据来自未见过的疾病状态,性能也会明显下滑。这意味着:AI 注释可以快,但不能偷懒。
五、AI会不会取代传统方法?
这篇综述给出的答案其实很冷静:
不会,至少现在不会。
在 scRNA-seq 的基础降维、聚类和批次校正里,DL 并没有稳定证明自己比成熟的传统方法更好。比如 scVI 的低维表示在独立 benchmark 里通常和 PCA 差不多;批次整合里,Harmony 依旧是非常强的基线,而不少 DL 方法反而容易出现过度校正,把真正的生物学差异也一起抹掉。
所以更准确的说法不是“AI替代了传统流程”,而是:
传统方法负责稳,AI负责灵活。
当你做的是常规 scRNA-seq 聚类、常规批次校正、常规 marker 分析,传统方法往往已经足够强;但当你需要同时吃下图像、空间、RNA、ATAC、蛋白这些异构数据时,AI 的价值才开始真正放大。
六、那生物学研究者到底该怎么用AI?
如果把这篇综述压缩成一句实用建议,那就是:
把AI当成增强器,不要当成裁判。
你可以让 AI 帮你做第一轮自动注释,帮你整合多组学,帮你融合空间图像和表达,帮你提取更复杂的表征;但你不能因为模型输出了一个更平滑的 UMAP、更漂亮的轨迹、更完整的表达矩阵,就默认它更接近真实 biology。真正决定结论能不能站住的,依然是实验设计、基准比较、人工复核和独立验证。
对今天的单细胞和空间转录组来说,AI 确实已经很重要,但它最可靠的角色还不是“终极判断者”,而是“强大的中间层工具”。它能帮我们更快地组织复杂数据、更方便地连接不同模态,也能把过去难以放到同一张图里的信息拼起来。
但从这篇综述的判断来看,AI最值得期待的价值,不是把传统方法全部替换掉,而是把过去根本拼不起来的数据世界,先拼起来。