 夜雨聆风
夜雨聆风





TCGA数据库使用指南:从下载到清洗入库,全程R代码公开!(附纯生信发文策略)

TCGA(The Cancer Genome Atlas),这个汇集了33种癌症类型、超过20000个样本的多组学“人类癌症百科全书”,从基因组、转录组到甲基化、蛋白质组,再到详尽的临床随访信息,几乎涵盖了肿瘤研究的全部维度。
你可以用它做单基因的全面扫描,也可以构建预后模型、描绘免疫微环境、开展泛癌分析,甚至联合多组学解释机制、预测药物敏感性。但纯数据挖掘早已不是“下载即发文”的坦途——外部验证、前沿热点、乃至干湿结合,才是让文章脱颖而出的关键。而这一切的起点,恰恰是从数据下载到清洗的每一步细节开始。
01
什么是TCGA数据库?
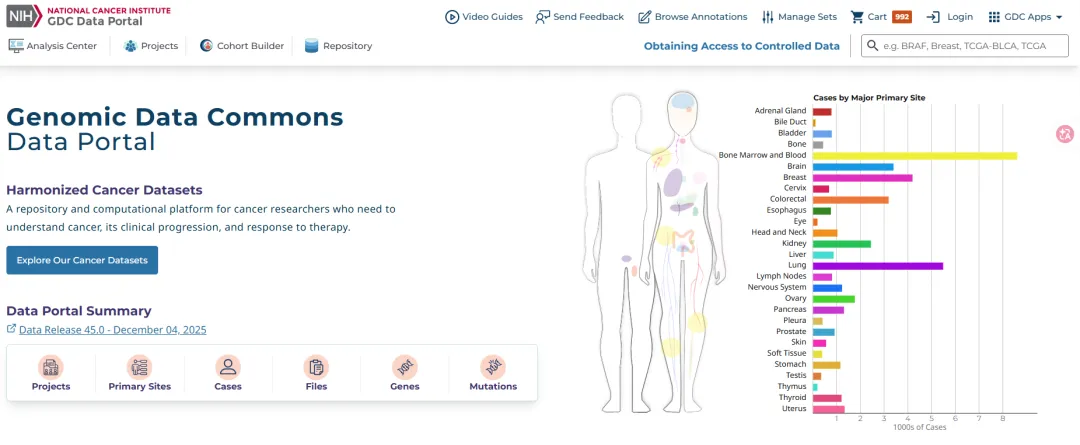
TCGA是由美国国家癌症研究所(NCI)等机构联合开展的项目。简单来说,它就是一个“人类癌症百科全书”。 它收录了33种癌症类型、超过20,000个样本的多组学数据和临床数据。在这里,你可以下载到(原始测序数据需授权,衍生矩阵数据可直接开放获取):
-
测序数据:基因组(WES/WGS)、转录组(RNA-Seq)、表观组(DNA甲基化)、蛋白质组(RPPA)等。
-
临床数据:患者性别、年龄、生存期(OS/DFS等)、肿瘤分期(TNM)、病理分级等。
02
基于TCGA的研究内容
1️⃣ 单基因/基因家族的全面生信分析
如果你在实验中偶然关注到一个基因,或者某个基因家族(如MMPs、N6-甲基腺苷(m6A)相关基因),你可以通过TCGA进行详细的分析:
-
表达差异:癌组织 vs 正常组织中该基因表达量的高低。
-
临床相关性:表达量与肿瘤分期、肿瘤转移有没有关系。
-
预后分析:分析该基因是保护因素,还是危险因素。
-
富集分析:做个GO/KEGG/GSEA分析,看看这个基因参与了什么信号通路。
2️⃣ 构建预后风险模型(Risk Signature)
单基因说服力不够,可以把多个基因打包在一起,构建一个评价模型。
-
结合当前热点:结合热点(如铁死亡、铜死亡、自噬、焦亡、代谢相关基因),在TCGA中找到与这些热点相关的差异基因。
-
结合机器学习筛选关键基因:利用LASSO回归、Cox回归等统计算法(推荐使用多种方法,避免出现方法上的偏倚),筛选出几个核心基因,构建一个评分计算公式(Risk Score)。
-
模型评估:把患者分为高低风险组,比较生存率,并运用ROC曲线证明模型预测能力超强。
3️⃣ 肿瘤免疫微环境(TME)与免疫浸润分析
肿瘤不是一座孤岛,它周围围绕着各种免疫细胞。
-
免疫细胞浸润的差异:利用ESTIMATE、CIBERSORT、ssGSEA等算法,把TCGA的RNA-seq数据转化为免疫细胞的丰度。分析目标基因/模型与哪种免疫细胞(如CD8+ T细胞、巨噬细胞)成正相关/负相关。
-
指导免疫治疗:不同分组对免疫检查点(PD-1, CTLA-4)的表达是否有差异?能否预测患者对免疫治疗(如PD-1抑制剂)的响应?也可以利用患者的免疫表型评分(IPS),预测不同类型患者对抗CTLA-4和抗PD-1抗体反应的差异。
4️⃣ 泛癌分析
只看一种癌症太局限?TCGA包含多种种癌症,我们可以把一个基因或预测模型在不同种类的癌症中进行评价。主要优势在于图表极其丰富(雷达图、环状热图等),给人一种“工作量巨大、视野广阔”的震撼感。
-
展示该基因或者在所有癌症中的突变图谱、表达模式、预后价值和免疫相关性。
-
预测模型在所有癌症中预后价值和免疫相关性。
5️⃣ 多组学联合分析
结合多维度数据,不仅能说明“现象”,还能解释部分“机制”。
-
转录组+甲基化:是不是因为启动子区低甲基化导致了基因高表达?
-
转录组+基因突变/CNV:基因突变与基因表达的关联。
-
转录组+蛋白组学:如果在转录组中发现mRNA高表达,在蛋白组中验证了蛋白也高表达,这就相当于做了一个大样本的“线上Western Blot(WB)”,同样具有一定的说服力。
6️⃣ 靶向药物敏感性预测
-
结合TCGA数据与GDSC(药物敏感性数据库)等外部资源。
-
分析你的目标基因高/低表达人群,对哪种化疗药物(如顺铂、紫杉醇)或靶向药物更敏感(计算IC50值)。这为临床个体化用药提供了理论依据。
由于纯挖TCGA的文章越来越多,现在单靠TCGA发SCI确实有难度,你需要掌握以下的“加分项”:
1.必须要外部验证:用TCGA作为分析分析对象,一定要去GEO数据库下载另外的独立队列来验证你的结论。如果两个数据库得出的结论一致,文章的可信度将大大增加。
2.结合当下前沿热点:结合最新的概念或者方法,如:空间转录组、单细胞测序(scRNA-seq)、各种新型细胞死亡方式、肠道菌群相关等。
3.补充基础的“湿实验”验证:纯“干实验”容易被质疑。如果你能在医院收集几对癌和癌旁组织,或者实验室有对应的肿瘤细胞系,做一下简单的RT-qPCR、WB或免疫组化(IHC),验证一下你的生信分析结果,或者验证关键基因的功能。
这样一来,不仅能让数据看着更扎实,课题的完整度也能提高,不至于让人觉得只是简单的“数据挖掘”。
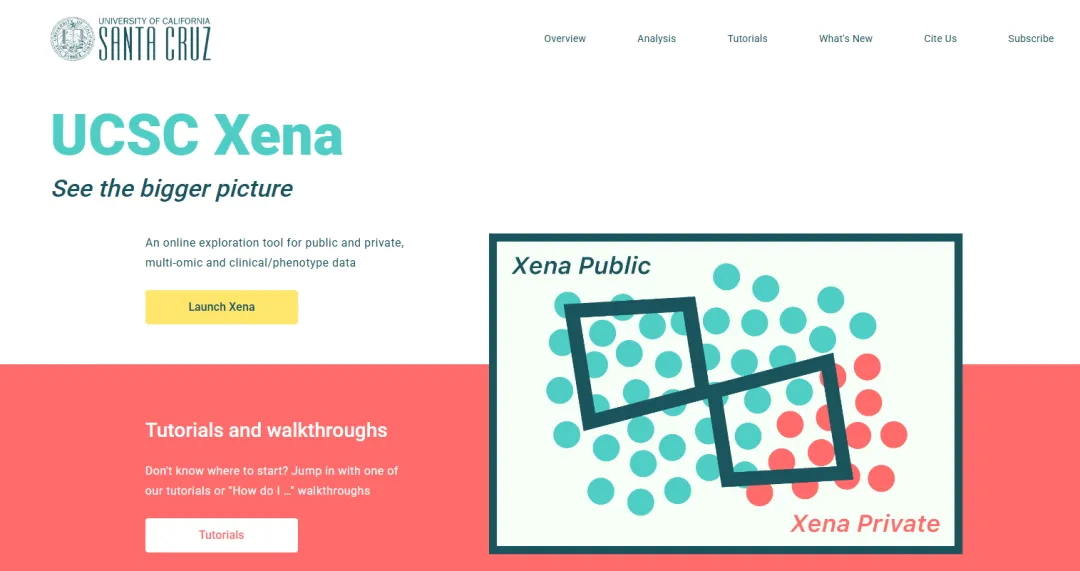
第一次接触TCGA时,面对这么多数据,大家可能觉无从下手。今天小编给大家介绍一个简单获取的数据平台,对我们的学习以及初步分析有很大的助力。
今天,我们就以UCSC Xena平台为例,手把手带你走一遍TCGA数据从获取到预处理的完整流程,让你的分析地基打得既稳又实。
-
UCSC Xena官网:https://xena.ucsc.edu/
点击“Launch Xena”启动网站,可以直接跳转进入可视化页面。
数据里包括整理好的TCGA中的各类癌症的数据,我们这里以ESCA为例:

以下就是ESCA队列中包含的数据, 我们这次主要分析RNAseq的数据。包含count以及两种标准化(TPM,FPKM)后的数据。
以TPM数据为例,列表中包含ID,样本数量等信息,并且提供了数据的下载链接,值得注意的是ID/Gene Mapping也需要下载下来,用于后续的ID转换。
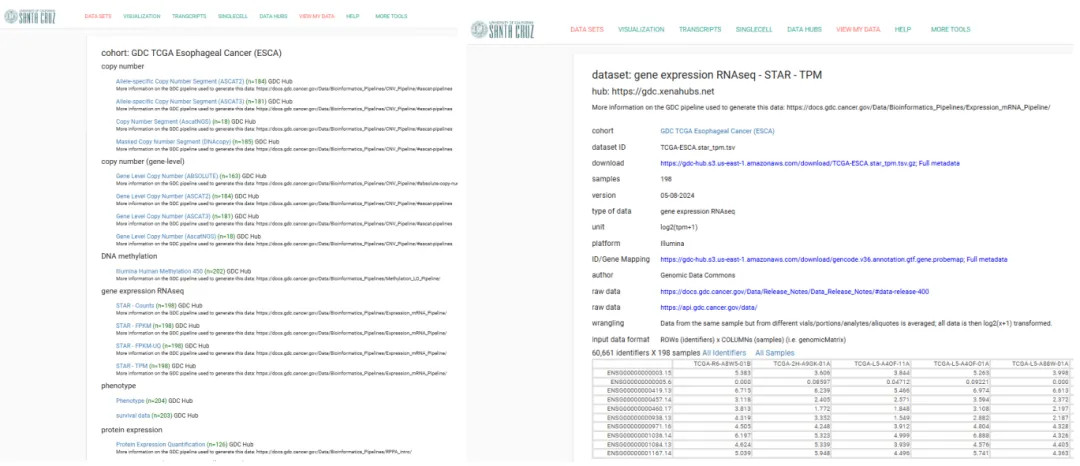
下载下来之后我们就可以用R来进行读取和预处理啦。
步骤1:读取数据

步骤2:提取基因ID与基因名对应关系

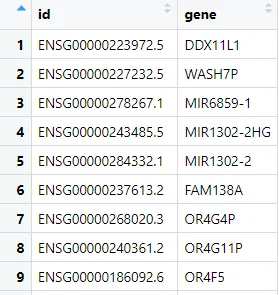
步骤3:将表达矩阵中的探针ID替换为基因名

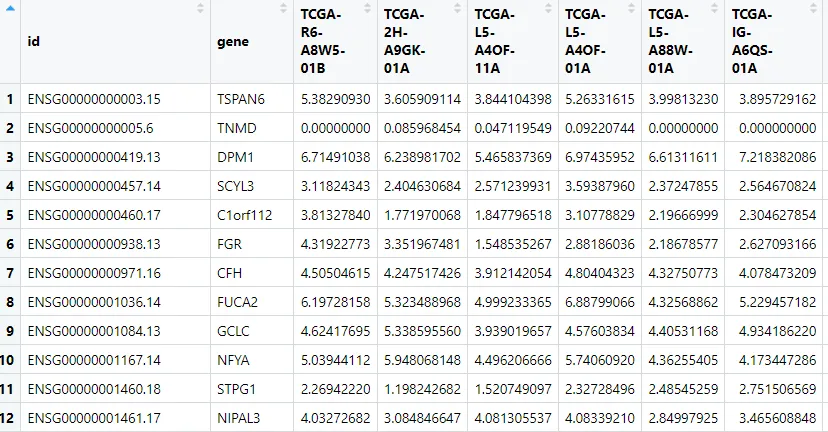
步骤4:处理多探针对应同一基因的去重
由于多个探针可能靶向同一个基因,我们需要对同一基因的多行进行去重。常用的策略是:计算重复行的均值。
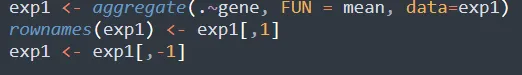
步骤5:提取临床信息与分组
这里比仅有正常组和疾病组的分组,同时还包含生存信息,肿瘤分期等临床信息。
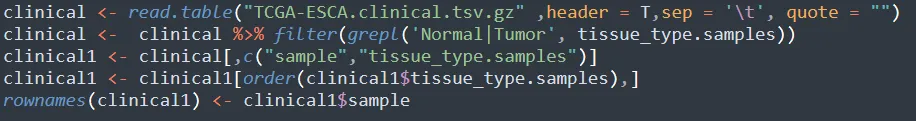
步骤6:提取同时具有分组信息和测序数据的样本,并保存测序数据
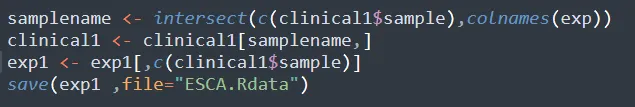
这样我们就既有表达量信息又有分析信息,可以进行进一步的分析了。
总 结
TCGA的价值,从来不限于它存储了多少组学信息,而在于我们如何用严谨的提问、合适的方法和充分的验证,把这些数字变成对肿瘤生物学有意义的洞察。
无论你选择纯生信挖掘作为快速探索的起点,还是最终走向干湿结合的纵深研究,请记住:每一次规范的下载、每一步仔细的去重和注释,都是在为结论的可信度添砖加瓦。

国内老牌的医学科研一对一培训平台,提供多种类型的科研与统计方法一对一指导,坚持授人以渔,助力医护工作者科研成长!
已指导1000+医学人掌握方法、成功发表SCI!

扫码联系业务 咨询详情👆

往期精彩
1
2
3