 夜雨聆风
夜雨聆风





零实验发一区!30万单细胞+AI虚拟敲除,炎癌转化直接起飞

从慢性炎症到癌症,究竟发生了什么?
全球约20%的癌症与慢性炎症直接相关:
· 乙肝/丙肝→肝癌
· 幽门螺杆菌感染→胃癌
· 炎症性肠病→结直肠癌
· Barrett食管→食管腺癌
炎症微环境如何一步步“叛变“、正常细胞如何逐渐“黑化“、哪个节点是阻断癌变的最佳窗口?这些问题的答案,直接关系到癌症的早筛、早诊和早治。
传统研究难以捕捉到疾病进展的动态过程
大多数研究只能比较“正常vs肿瘤“两个端点,中间的炎症、癌前病变、早期癌变等关键阶段往往被忽略。更棘手的是,即使拿到了不同阶段的样本,但往往不是配对患者的数据。如何解决数据的批次问题,识别驱动进展的关键基因、验证它们的功能,仍然有待解决。
今天的这篇文章,给出了一套全新的解法

研究者整合了6个公开数据集的30万+单细胞,构建了从“正常肝→原发肿瘤→门静脉癌栓→淋巴结转移“的完整炎癌转化图谱;随后用Geneformer大语言模型进行虚拟基因敲除——让AI”想象“敲掉某个基因后细胞会发生什么变化——最终锁定了贯穿疾病全程的治疗靶点HSP90B1。
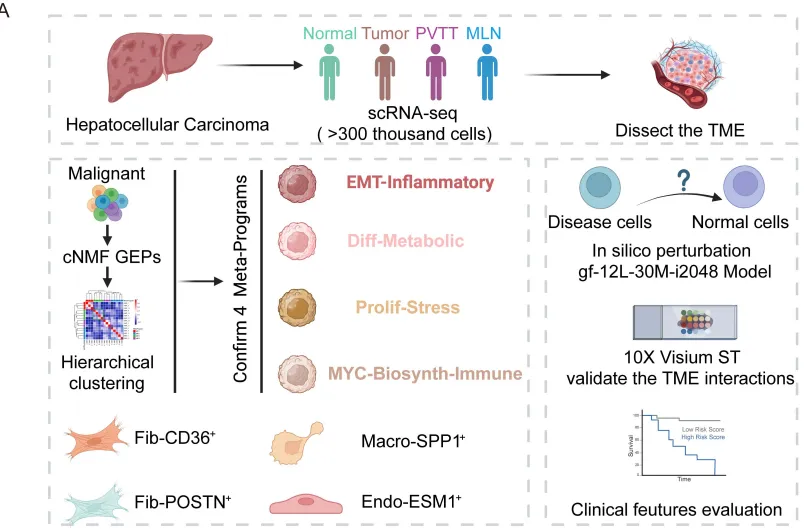
这套“公共数据+AI虚拟扰动“的组合拳,正在重新定义炎癌转化研究的玩法:
· 不需要自己测序,整合公共数据就能构建大规模疾病图谱
· 不需要做敲除实验,AI虚拟扰动就能预筛候选靶点
· 不需要盲目验证,多维度数据交叉验证确保可靠性
下面我们来拆解这篇文章的核心发现,以及如何把这套方法用到你自己的课题里。
想用公共数据发一区?
扫码获取动态进展+虚拟扰动研究方案

多组学与AI驱动下的疾病进展机制探究
用公共数据构建“疾病进展的连续快照”
研究者整合了6个GEO数据集(GSE125449、GSE149614、GSE151530、GSE156625、GSE134355、GSE189903),涵盖正常肝→原发肿瘤→门静脉癌栓→淋巴结转移的完整疾病谱系,质控后保留115个样本、超过30万个细胞。
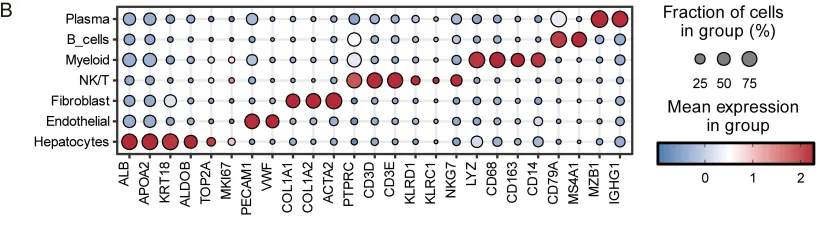
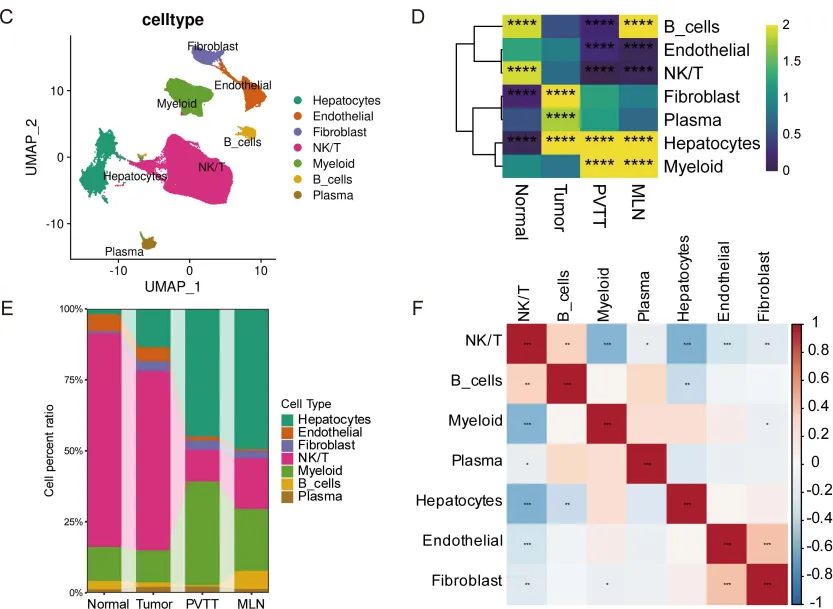
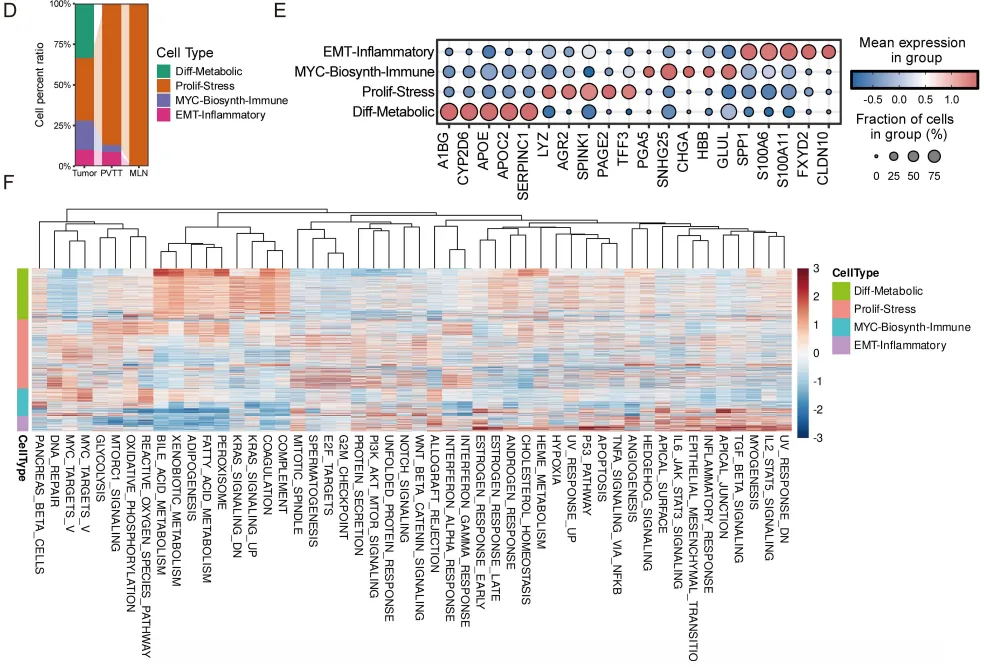
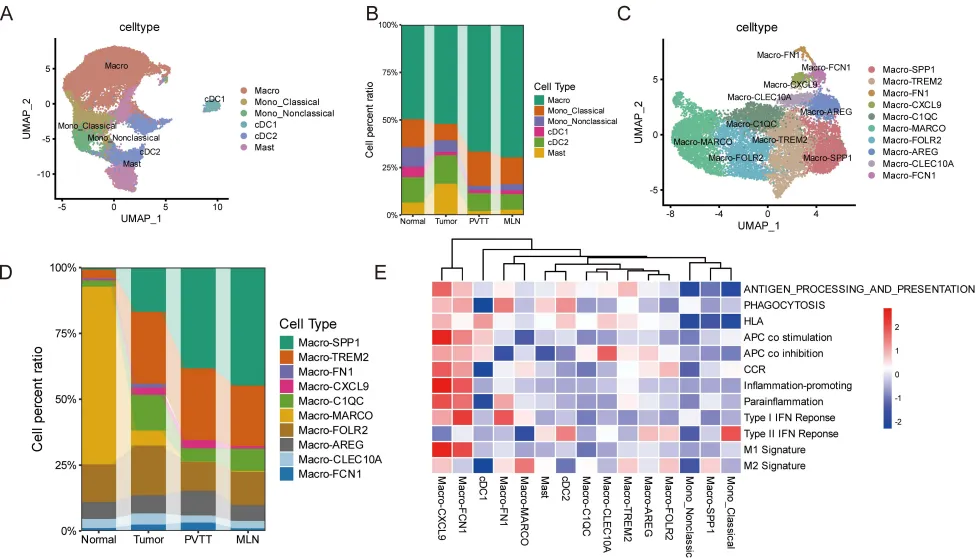
第一个发现:恶性细胞的“四种命运”
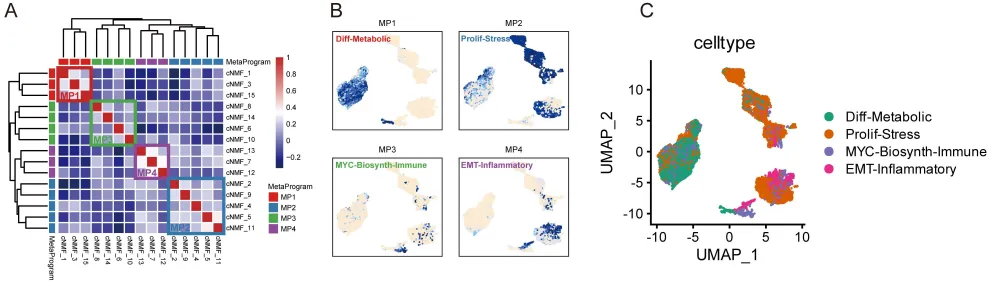
通过cNMF(共识非负矩阵分解)分析,研究者在恶性肝细胞中识别出四种转录状态,每种状态代表一种不同的“细胞命运“:
· Diff-Metabolic:保留肝细胞代谢功能,MYC/E2F被抑制,预后较好,但对索拉非尼耐药
· Prolif-Stress:高增殖+应激反应,MYC/E2F激活,预后差,对HSP90抑制剂敏感
· MYC-Biosynth-Immune:MYC驱动的生物合成+先天免疫重编程,预后差
· EMT-Inflammatory:EMT+TGFβ+炎症信号,预后最差,对MEK抑制剂敏感
这四种状态不是随机分布的。原发肿瘤中四种状态均衡存在,但到了PVTT和MLN,Prolif-Stress和EMT-Inflammatory细胞占据主导。特别是淋巴结转移灶,几乎被Prolif-Stress细胞“垄断“——这提示高增殖细胞具有选择性转移优势。
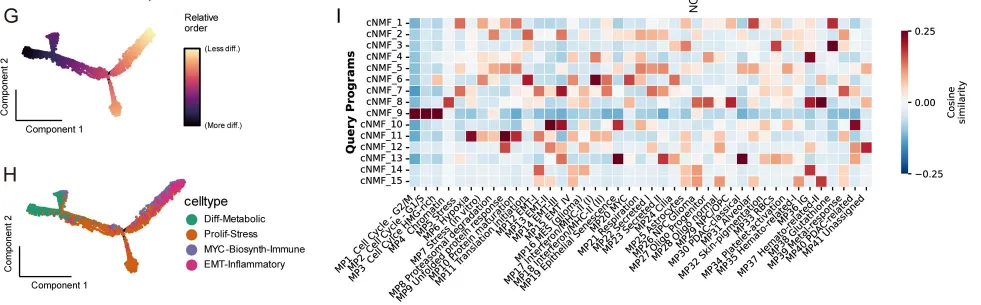
拟时序分析进一步揭示:EMT-Inflammatory细胞位于轨迹起点,向Diff-Metabolic和Prolif-Stress两个方向分化。这意味着EMT-Inflammatory可能是肿瘤演化的“源头状态“。
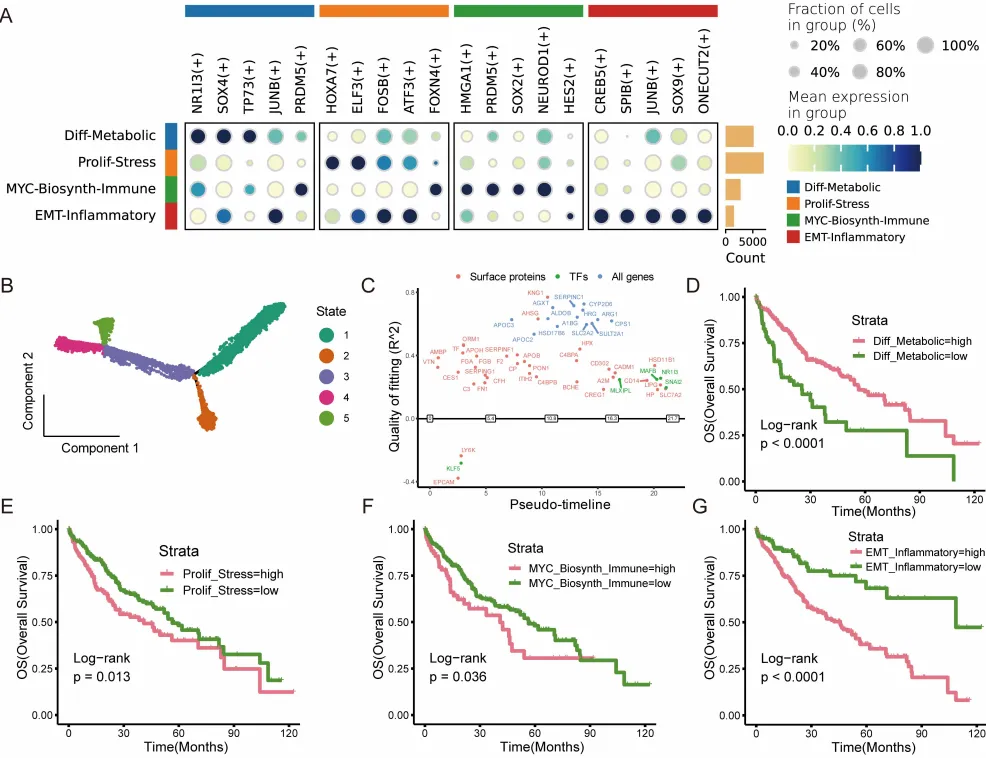
研究者通过pySCENIC分析揭示了各状态的核心调控网络:
· Diff-Metabolic细胞由肝细胞谱系转录因子(NR1I3、SOX4、TP73)主导,这解释了它们为何保留代谢功能
· Prolif-Stress细胞由细胞周期调控因子(HOXA7、ELF3)驱动
· EMT-Inflammatory细胞由间充质转录因子(CREB5、JUNB、SOX9调控
值得注意的是,部分转录因子在多个状态间共享——如JUNB同时活跃于Diff-Metabolic和EMT-Inflammatory细胞,ATF3和FOSB同时富集于Prolif-Stress和EMT-Inflammatory细胞。这种“调控网络的部分重叠“可能是肿瘤细胞保持表型可塑性、实现状态转换的分子基础。
研究者在TCGA-LIHC队列中进行了验证:
高Diff-Metabolic评分与较好的总生存期相关,高Prolif-Stress、MYC-Biosynth-Immune、EMT-Inflammatory评分均与较差预后相关
更有意思的是索拉非尼反应分析:所有四种状态在非应答者中评分都更高,其中Diff-Metabolic在非应答组中富集最明显。这看似矛盾——预后最好的状态,为何对药物最不敏感?
文中给出的解释是:Diff-Metabolic细胞保留了肝细胞的药物代谢能力,其核心调控因子NR1I3(又称PXR)是异生物质代谢的主调控器,可能将索拉非尼转化为无活性代谢物。分化良好≠治疗敏感,这一发现对临床用药策略有重要启示。
第二个发现:微环境的重塑
肿瘤细胞不是孤军奋战,它们需要微环境的“配合“。研究者发现,随着疾病进展,微环境经历了系统性重塑:
· 巨噬细胞的“叛变“:正常肝脏中的Kupffer细胞(Macro-MARCO)逐渐被免疫抑制性的Macro-SPP1和Macro-TREM2取代。Macro-SPP1尤其“恶劣“——它富集于血管生成、EMT、糖酵解和缺氧通路,同时干扰素-γ反应被抑制。
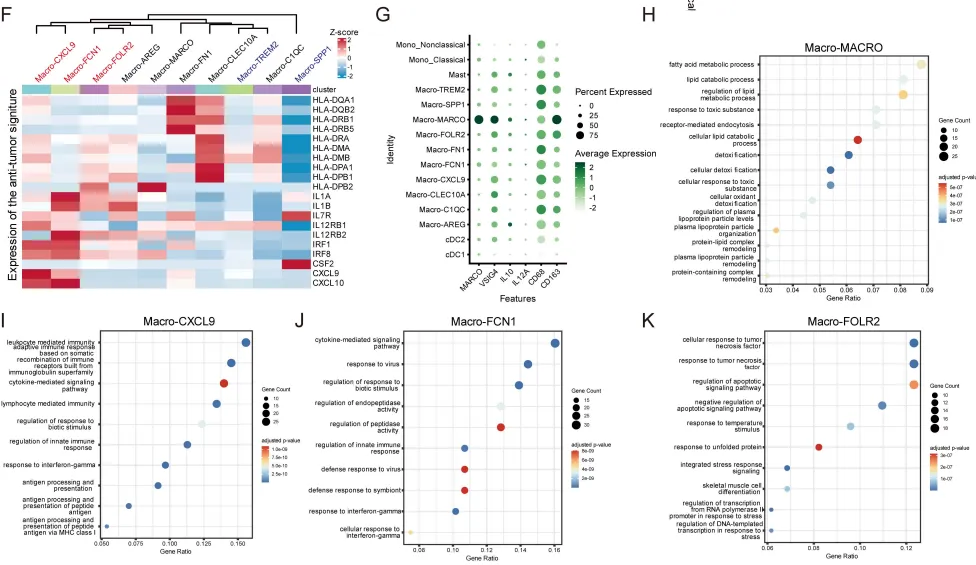
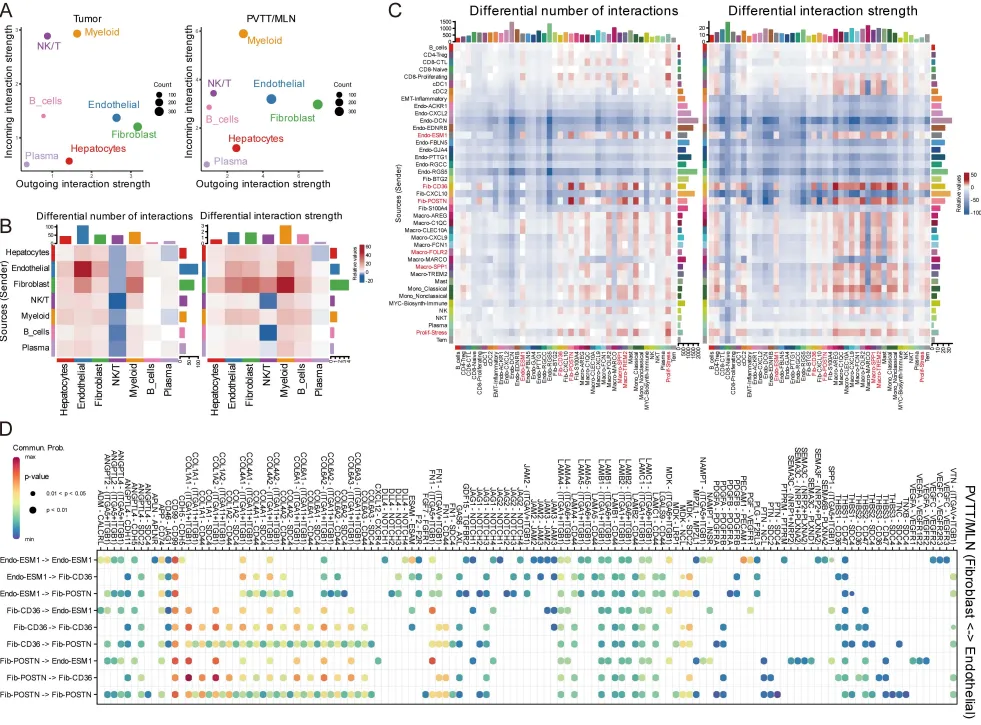
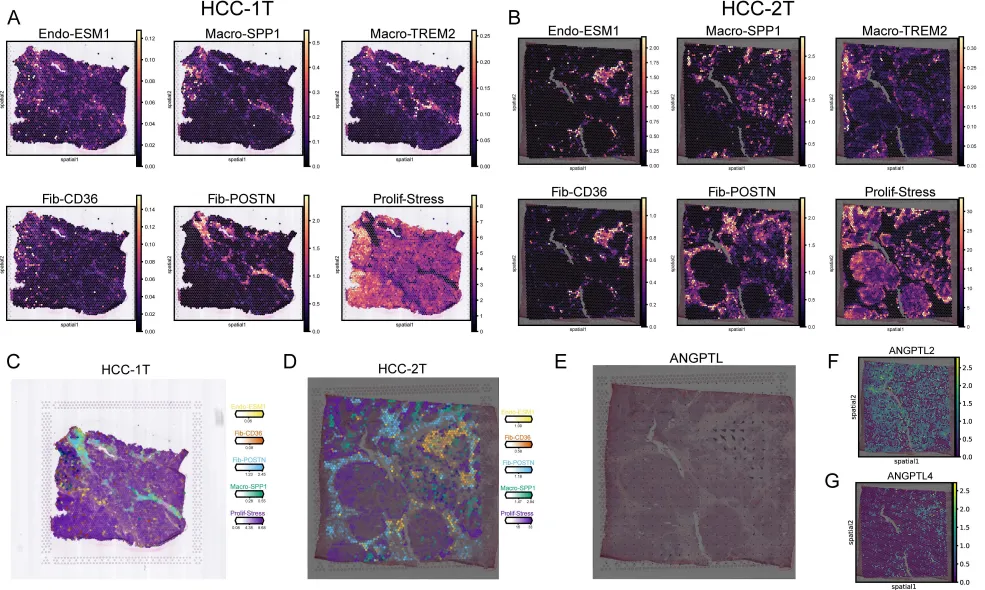
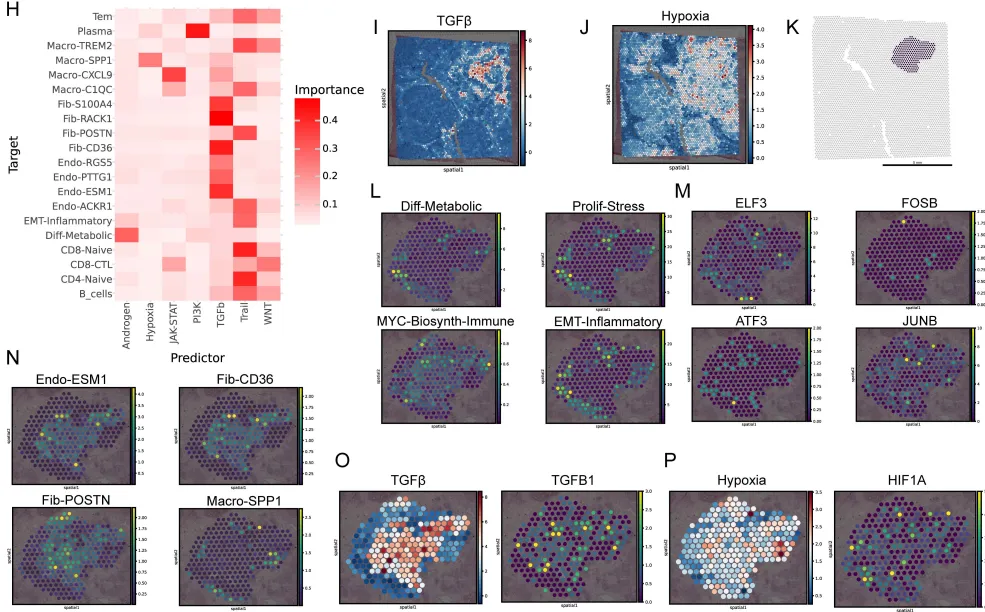
空间转录组验证:这种内皮–成纤维细胞联盟不是随机分布的,而是形成有组织的“基质核心“,周围环绕着Prolif-Stress和EMT-Inflammatory肿瘤细胞。TGFβ信号在基质核心高度富集,缺氧信号则弥漫于整个区域。
这揭示了一个层次化的微环境架构:中央基质核心(内皮+成纤维细胞)→TGFβ富集区→外周肿瘤细胞(Prolif-Stress/EMT-Inflammatory)→缺氧区。
Geneformer虚拟扰动——不做实验也能”敲基因”
传统靶点筛选需要大量实验验证,耗时耗力。研究的另一大亮点是使用Geneformer——一个在约3000万单细胞转录组上预训练的大语言模型——进行虚拟基因敲除。
什么是虚拟扰动?简单说,就是让AI”想象“:如果敲除某个基因,细胞的转录组会发生什么变化?细胞状态会如何转换?
研究者设计了三个细胞状态转换:
· 将免疫抑制性Macro-SPP1→免疫激活性Macro-CXCL9
· 将恶性肝细胞→正常肝细胞
· 将病理性Endo-ESM1→正常Endo-EDNRB
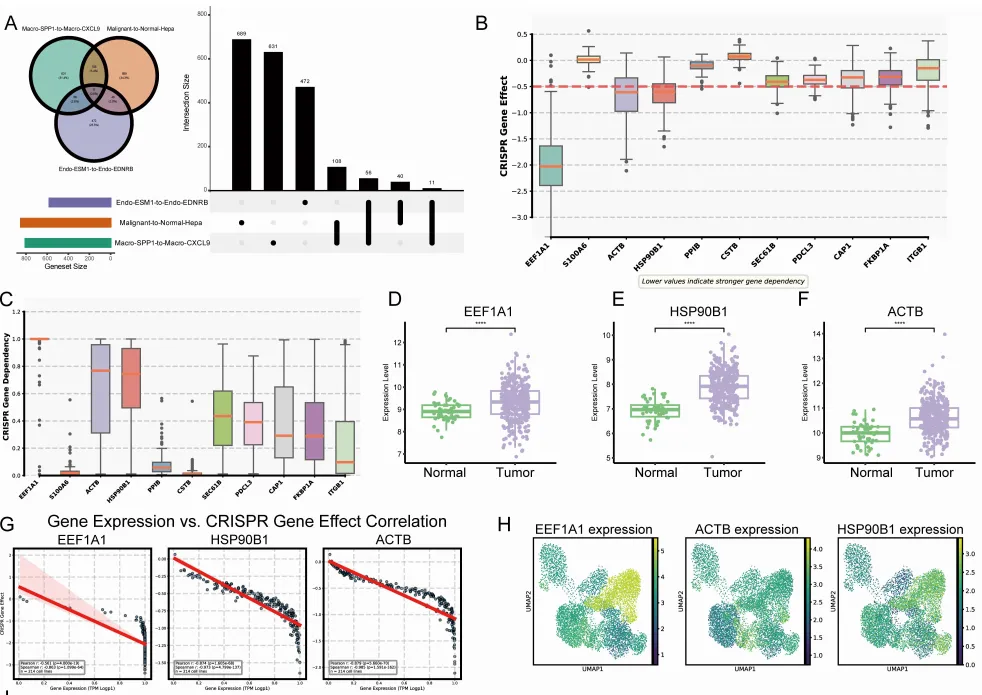
结果发现,11个基因在所有三种转换中被共同识别,其中HSP90B1脱颖而出:
· DepMap数据库显示其在癌细胞中具有强依赖性
· TCGA-LIHC显示其在肿瘤中显著上调
· 表达–依赖性相关系数高达-0.874(表达越高,细胞越依赖它)
· 在索拉非尼和cabozantinib+nivolumab治疗的非应答者中均显著升高
· 高表达与不良预后显著相关
进一步分析了HSP90B1在治疗应答者和非应答者中的空间分布模式:
· 应答者:HSP90B1呈局灶性分布,集中在特定区域
· 非应答者:HSP90B1呈弥漫性分布,遍布整个肿瘤组织
这种空间分布差异提示:HSP90B1的空间表达模式可能成为预测治疗反应的新型标志物。局灶性分布可能意味着肿瘤对HSP90B1的依赖是局部的、可控的;而弥漫性分布则提示全面的依赖和更强的耐药潜力。
这套范式如何应用于其他动态进展研究?
· 在各种器官纤维化(如肝、肺、肾、心)的进程中,组织会经历从急性损伤、炎症浸润、到基质细胞异常活化、最终导致不可逆纤维化和器官衰竭的明确阶段。应用此范式,我们可以系统追踪究竟是哪一群成纤维细胞前体被异常激活,又是哪些来自受损上皮或慢性免疫细胞的信号(如TGF-β、WNT)在持续驱动这一过程。通过虚拟扰动,探究扰动哪些靶点可以将终末期致密的肌成纤维细胞逆转为静息状态?这为发现不仅能阻止、更能逆转纤维化的革命性疗法提供了全新的发现路径。
· 在急性损伤与修复中(如脑卒中、心肌梗死、急性肾损伤),组织经历损伤、炎症、修复/再生的经典病理过程,但修复失调会导致慢性纤维化与功能障碍。利用此框架,我们可以精细解析在修复早期,哪些促再生信号被激活,而在哪些患者或情况下这些信号会过早关闭,转而启动促纤维化程序。通过比较恢复良好与恢复不良患者的单细胞图谱,结合空间技术定位“再生微环境”与“瘢痕微环境”的差异,虚拟扰动模型可以帮助我们筛选出能强化内源性修复程序、抑制异常疤痕形成的关键分子开关,从而将治疗窗口从单纯的急性期保护,延伸到促进长期功能性恢复。
虚拟扰动:不只有Geneformer
除了本文使用的Geneformer,还有多种虚拟扰动工具可供选择:
· scTenifoldKnk:基于张量分解的虚拟敲除工具,可以模拟单基因敲除后转录组的变化,已被用于结直肠癌CD4+ T细胞靶点筛选。
· CellOracle:基于基因调控网络的扰动预测工具,可以模拟转录因子敲除/过表达的效果,发表于Nature。
· GEARS:基于图神经网络的扰动预测模型,可以预测组合扰动的效果,发表于Nature Biotechnology。
· scGen:基于变分自编码器的扰动预测工具,可以预测药物处理后的细胞状态变化。
· Pertpy:发表于Nature Methods的Python框架,专门针对单细胞扰动数据的分析需求进行了全方位设计。
这些工具的共同理念是:在实验之前,先用计算模拟“预演”——预测哪些基因值得敲除、哪些药物可能有效、哪些组合值得尝试。这大大降低了实验成本,加速了靶点发现。
想用公共数据发一区?
扫码获取动态进展+虚拟扰动研究方案

多组学与AI驱动下的疾病进展机制探究
参考文献:
1.Xia P, et al. Large-scale single-cell analysis and in silico perturbation reveal dynamic evolution of HCC: from initiation to therapeutic targeting. npj Precision Oncology, 2026. https://doi.org/10.1038/s41698-026-01307-2
2.Osorio D, et al. scTenifoldKnk: An efficient virtual knockout tool for gene function predictions via single-cell gene regulatory network perturbation. Patterns, 2022. https://doi.org/10.1016/j.patter.2022.100434
3.Kamimoto K, et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature, 2023. https://doi.org/10.1038/s41586-022-05688-9
4.Roohani Y, et al. Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nature Biotechnology, 2024. https://doi.org/10.1038/s41587-023-01905-6
5.Lotfollahi M, et al. scGen predicts single-cell perturbation responses. Nature Methods, 2019. https://doi.org/10.1038/s41592-019-0494-8
6.Palla G, et al. Pertpy: an end-to-end framework for perturbation analysis. Nature Methods, 2024. https://doi.org/10.1038/s41592-024-02233-6
7.Theodoris CV, et al. Transfer learning enables predictions in network biology. Nature, 2023. https://doi.org/10.1038/s41586-023-06139-9
热点思路推荐
|
|
|
|
|
|
|
|
|
|
|
|
临床医生发文指南
