 夜雨聆风
夜雨聆风





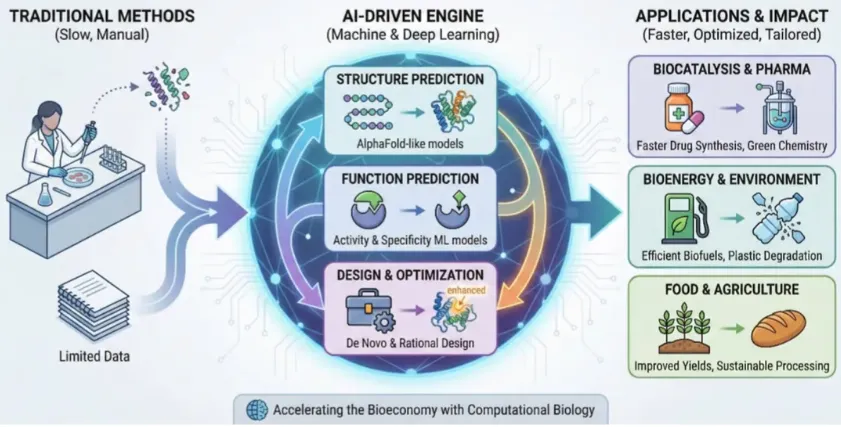
轰动全球!AI驱动的酶工程强势破局:从结构预测到全新设计,太逆天!
八大热门专题


MONDAY


01、人工智能构建虚拟细胞

02、AI蛋白质设计

03、AI抗菌肽设计

04、CADD计算机辅助药物设计

05、AIDD人工智能药物发现与设计

06、合成生物学与基因电路设计

07、机器学习代谢组学

08、深度学习在多组学融合中的应用

特惠福利:
(报名一门直播课赠送两门回放)
(报名三门直播课赠送以下全部)
01.单细胞测序与空间转录组学
02.深度学习解析宏基因组学
03.机器学表观组与转录组
04.深度学习质谱蛋白组学
05.机器学习生物医学
06.AI智慧医疗影像
07.医疗AI多模态大模型
08、深度学习在基因组学中的应用
09、CRISPR-Cas9基因编辑技术
10、机器学习在微生物组学中的应用
11、CADD(进阶班)


上午 理论讲解(第三、四阶段)
第三阶段:细胞调控机制建模(Regulatory Modeling)
核心目标:解决“为什么细胞会发生变化”,讲解细胞调控的底层机制,从表型识别深入到机制层面,明 确调控机制建模在药物研发中的核心价值。
配套模型理论:细胞调控网络与机制推理模型(Regulatory Network Models),详细讲解Graph Attention Network (GAT)、Message Passing Neural Network (MPNN)、SCENIC、Gene Regulatory Graph、Protein Interaction Graph 的核心原理,阐述其核心结果(TF调控网络、基因因 果关系、蛋白互作机制),以及在药物靶点发现中的应用逻辑。
第四阶段:细胞动态预测(Dynamic Evolution)
核心目标:解决“细胞下一步会走向哪里”,讲解细胞命运轨迹推演的核心逻辑,以及动态预测对药物研 发(如耐药、复发预测)的重要意义。
配套模型理论:结合细胞状态建模与调控机制理论,补充细胞动态预测的核心思路,衔接大模型驱动的细 胞语言模型基础,为下午scGPT、CellRank 2实操铺垫理论基础。
下午 实操演练(对应上午第三、四阶段理论)
实操前置准备:图神经网络与动态预测工具包调试
1. Python 工具包适配:PyTorch Geometric(图神经网络)、CellRank(动态预测)工具包的安装与调试;
2. 数据预处理复习:回顾上午理论相关的基因表达数据、调控关系数据的预处理方法。
实操模型讲解(Python代码解析 + GPU服务器上机实操)
1. 实操模型3:GAT(调控网络机制推理)—— 对应第三阶段理论,基于基因表达数据,构建基因调控 网络,识别关键调控节点,掌握机制推理的核心方法,理解其在药物靶点发现中的应用;
2. 实操模型5:CellRank 2(命运与轨迹推演)—— 对应第四阶段理论,基于单细胞数据,推演细胞分 化轨迹,预测细胞未来状态,掌握动态预测的核心方法,贴合药物研发中耐药、复发预测的需求;
3. 实操模型4:scGPT(基础大模型时代)—— 衔接第四阶段动态预测理论,基于基因表达数据,完成 细胞语言特征提取,预测药物扰动前后的细胞状态差异,掌握大模型在细胞动态预测中的基础应用。
上午 理论讲解(第五阶段)
第五阶段:药物作用建模(Drug Perturbation Modeling)
核心目标:解决“药物如何改变细胞命运”,讲解药物作用于细胞的核心逻辑,以及药物扰动建模在药物 研发全流程中的应用场景(筛选、联合用药、毒性预警等)。
配套模型理论:生成式药物扰动虚拟细胞模型(Generative Perturbation Models),详细讲解 scDiffusion、Geneformer perturbation mode、CPA、ChemCPA、scGen、CellOT 的核心原理,明 确其核心结果(单药作用、联合给药、剂量反应、细胞未来状态预测),以及在药物筛选预实验、联合用 药预测、毒性预警、MOA机制解释中的具体应用。
下午 实操演练
实操前置准备:药物扰动模型工具包调试
1. Python 工具包适配:ChemCPA、scGen等药物扰动相关工具包的安装与调试;
2. 数据准备:药物作用相关数据(药物剂量、细胞反应数据)的预处理与导入方法。
实操模型讲解(Python代码解析 + GPU服务器上机实操)
1. 实操模型6:ChemCPA(药物扰动预测)—— 对应第五阶段理论,构建药物扰动模型,预测不同药物 剂量的作用效果、联合用药反应,掌握虚拟筛选的核心能力,理解其在药物研发ROI提升中的作用;
2. 补充实操:结合scGPT模型,预测药物扰动前后的细胞语言差异,衔接大模型与药物扰动建模的核心 逻辑,巩固上午理论与实操的联结。


上下滑动查看更多


学习目标: 理解现代蛋白质设计的理论基石,建立从“功能”到“结构”再到“序列”的正确设计观。
蛋白质折叠问题与经典设计:
。回顾“CASP历史”以及蛋白质折叠问题的复杂性 。
。介绍David Baker实验室与IPD的设计哲学:以最终功能为导向,设计自然界不存在的全新蛋白质结构。
。讲解Rosetta软件的核心思想,理解其作为蛋白质设计基石的历史地位与方法论
模块二:核心AI模型原理与代码实现
学习目标:从原理和代码层面,彻底理解驱动现代蛋白质预测与设计的两大核心神经网络架构。
·基础工具与环境:
o掌握Numpy进行多维数组(Tensor)操作 。
o学习PyTorch的核心概念:张量、自动求导机制
o讲解服务器GPU与CUDA的基本工作原理,为后续的本地化部署打下基础。
·关键模型架构拆解:
oTransformer与Attention机制:深入讲解Transformer模型如何通过自注意力机制捕捉序列中的长距离依赖关系,并分析其如何被AlphaFold 2用于整合多序列比对(MSA)信息和空间几何信息,构成Evoformer模块的核心 。
o扩散模型 (Diffusion Model):详细阐述扩散模型的前向加噪(Forward Process)与反向去噪(Reverse Process)的数学原理 。重点讲解该模型如何被应用于RFdiffusion,实现从高斯噪声中逐步生成结构合理的蛋白质骨架
代码实践:所有模型原理都将配合Jupyter Notebook进行代码实操,确保学员不仅理解理论,更能动手实现。
模块三:前沿设计工具链:原理、部署与应用
学习目标:掌握当前最高效的蛋白质设计“三要素”工具链,并完成本地化部署,具备独立开展计算设计的能力。
A. 结构生成 (RFdiffusion):
功能详解:学习RFdiffusion如何从随机噪声中生成全新的蛋白质骨架 。
实操技术:重点讲解几种核心设计模式的应用场景与参数设置:
无条件生成 (Unconditional generation):用于创造全新的拓扑结构。
基于骨架的生成 (Scaffolding):在已有的结构骨架上进行延申或构建。
功能位点限定生成 (Inpainting):在固定关键功能位点(如活性中心、结合界面)的前提下,生成包裹该位点的全新结构。
B. 序列设计 (ProteinMPNN):
o核心问题:讲解“蛋白质反向折叠 (Inverse Folding)”问题的挑战性,即如何为给定的骨架设计出能正确折叠的氨基酸序列 。
o协同工作流:演示如何将RFdiffusion生成的骨架(Backbone)作为输入,利用ProteinMPNN设计出高稳定性的氨基酸序列,形成完整的“结构-序列”设计流程 。
C. 结构验证与功能拓展 (AlphaFold 3):
o精度验证:学习使用AlphaFold 2/3验证由ProteinMPNN设计的序列,能否高精度地折叠回RFdiffusion所设计的初始结构 。重点讲解pLDDT和PAE图的解读,用于评估预测结果的置信度 。
o功能预测:介绍AlphaFold 3的核心突破——其预测能力已从单一蛋白质扩展到蛋白质、DNA、RNA、配体等多种分子的复合物 。探讨如何利用这一特性,初步评估设计的蛋白质与靶点分子的相互作用,为药物设计提供关键信息。
模块四:应用方向专题实战
学习目标:将前面学习的工具链应用到三个主流的生物医药研发场景中,解决具体问题。
总学习目标:通过三个独立的、项目驱动的专题,让学员完整地、端到端地执行计算蛋白质设计的流程。在课程结束时,学员不仅掌握了工具的使用,更能理解在不同应用场景下(酶、抗体、从头设计)的设计策略、关键考量和技术组合。
专题一:从头设计功能性Binder
技术流程:一个完整的综合性项目。学员将选择一个具体靶点,独立运用“RFdiffusion → ProteinMPNN → AlphaFold”的全套技术流程,从零开始设计一个能够特异性结合该靶点的全新小分子蛋白(Binder),完成一个标准化的de novo设计流程。
项目目标:执行一个完整的、工业界标准的de novo设计流程,针对一个选定的蛋白靶点,从零开始设计一个能够特异性结合其表面的全新小分子蛋白(Binder)。
核心概念:De novo设计、蛋白质-蛋白质相互作用(PPI)、形状互补性、计算流程的端到端整合。
技术流程详解:
1.靶点分析与结合位点选择:
o选择一个感兴趣的靶点蛋白(如疾病相关蛋白)。
o分析其表面性质,选择一个适合作为结合靶点的区域(通常是相对平坦、具有一定疏水性的表面)。
2.基于靶点的骨架生成 (RFdiffusion):
o此为设计的核心创造步骤。学员将学习使用RFdiffusion的功能位点限定生成 (Inpainting)模式。
o将靶点蛋白的结构作为固定的“环境”,RFdiffusion会在此环境的约束下,从噪声中“生长”出一个与靶点表面形状高度互补的全新蛋白质骨架。
3.序列填充与优化 (ProteinMPNN):
o将上一步生成的最优骨架输入ProteinMPNN。
oProteinMPNN会为这个全新的骨架设计出能够稳定折叠的氨基酸序列,完成从“形状”到“化学本质”的转化 。
4.双重计算验证 (AlphaFold):
o步骤一:单体验证。将ProteinMPNN设计的序列输入AlphaFold,预测其单体结构。验证该序列能否独立折叠成我们设计的形状(高pLDDT得分,且RMSD与设计模型低)。
o步骤二:复合物验证。使用AlphaFold-Multimer 预测设计的Binder和靶点蛋白的复合物。验证Binder能否如预期般结合到靶点的正确位置(低界面PAE得分)。
项目目标:针对一个给定的酶,通过计算方法,设计出具有更高热稳定性或催化活性的突变体,并提供一套可供实验验证的候选序列。
核心概念:适应性景观 (Fitness Landscape) ,定向进化 ,酶催化与过渡态理论 (Transition State Theory) ,计算诱变 (In-silico Mutagenesis),折叠自由能 (ΔG)。
技术流程详解:
1.模板选择与结构准备:
o从蛋白质结构数据库(PDB)中选取一个具有已知结构和功能的酶作为改造模板。
o若无实验结构,将学习利用UniProt 寻找同源序列,并使用SWISS-MODEL 或本地AlphaFold进行结构预测,获得可靠的起始模型。
o使用PyMOL 或ChimeraX 分析酶的活性位点、底物结合口袋以及潜在的不稳定区域(如高柔性loop)。
2.计算突变扫描与稳定性预测:
o学习使用Rosetta等工具包中的能量函数,对酶的特定区域(或全长)进行计算饱和突变扫描。
o核心技术点是计算每个突变体相对于野生型的折叠自由能变化 (ΔG)。学员将学习如何解读ΔG值,以筛选出能够提升蛋白质热稳定性的有益突变。
3.催化性能的计算评估:
o对于活性位点附近的突变,将引入分子对接(Molecular Docking)方法,模拟底物或过渡态类似物与突变酶的结合情况。
o通过比较结合能、关键相互作用(氢键、盐桥等)的变化,来半定量地预测突变对催化活性(kcat/KM)的潜在影响。
4.候选突变体的筛选与组合:
o基于上述计算结果,学员将学习如何制定筛选策略,例如,优先选择同时提升稳定性且不损害(或提升)活性的突变。
o探讨如何组合多个有益的单点突变,以获得性能提升更显著的多点突变体。
项目目标:掌握计算辅助的抗体亲和力成熟(Affinity Maturation)流程,针对一个已知的抗体-抗原复合物,设计新的互补决定区(CDR)序列,以增强其结合能力。
核心概念:抗体结构域、CDR与框架区(FR)、抗原表位(Epitope)与互补位(Paratope)、抗体人源化、亲和力与特异性。
技术流程详解:
1.抗体-抗原复合物的结构分析:
o从PDB中选取一个抗体-抗原复合物结构作为起始点。
o利用可视化软件,精确识别构成结合界面的CDR loop,特别是贡献最大的CDR-H3。
o详细分析界面上的氢键、疏水作用、盐桥等关键相互作用,理解亲和力的结构基础。
2.CDR区域的序列设计:
o固定抗体的框架区和抗原结构,仅针对CDR区域进行序列设计。
o学员将使用ProteinMPNN等工具,在保持CDR loop骨架构象的同时,重新设计其氨基酸序列,以期发现能与抗原形成更优相互作用的新序列。
3.利用AlphaFold-Multimer进行复合物结构预测与评估:
o将新设计的CDR序列整合回抗体,形成一个完整的突变抗体序列。
o使用AlphaFold-Multimer 预测新设计的抗体与抗原的复合物结构。这是流程中最关键的一步。
o学员将学习如何解读AlphaFold-Multimer的输出结果,特别是利用PAE(Predicted Aligned Error)图来评估界面预测的置信度,判断新设计的抗体是否仍能以正确的模式结合抗原。
4.亲和力排序与候选筛选:
o通过比较不同设计方案的界面结合面积、预测的氢键网络、以及界面PAE得分等指标,对设计的抗体进行排序。
o筛选出1-3个预测结合能力最强、且结构最可靠的候选抗体序列。


上下滑动查看更多


核心目标:掌握抗菌肽(AMP)的物理化学法则,并在云端建立可复现的数据集。
上午:AMP 定义理论与数据库实战 (3h)
1.AMP 设计的物理化学法则:
o机制:膜透化机制(Pore-forming)与螺旋轮图(Helical Wheel)投影分析。
o关键指标:如何计算两亲性力矩(Hydrophobic Moment)、净电荷与疏水性的平衡。
下午:Google Colab 基础与 Biopython (3h)
1.云端环境配置:
oGoogle Colab 显卡分配、挂载 Google Drive 数据持久化。
oJupyter 魔法命令 (!pip install, %cd) 的使用。
2.Biopython 序列处理:
o利用 Bio.SeqUtils.ProtParam 批量计算 5000 条序列的等电点 (pI) 和不稳定系数。
可视化:使用 seaborn 绘制正负样本的理化性质分布图(为后续条件生成做准备)。
参考文献 (Confirmed):
·[数据库] Wang, G., et al. (2016). “APD3: the antimicrobial peptide database as a tool for research and education.” Nucleic Acids Research, 44(D1), D1087-D1093.
·[去冗余] Fu, L., et al. (2012). “CD-HIT: accelerated for clustering the next generation sequencing data.” Bioinformatics, 28(23), 3150-3152.
·[理化性质] Eisenberg, D., et al. (1982). “The helical hydrophobic moment: a measure of the amphiphilicity of a helix.” Nature, 299(5881), 371-374.
蛋白质大模型与特征工程 (Protein LLMs & Feature Engineering)
核心目标:利用 Hugging Face 调用 Meta 的 ESM-2 模型提取序列特征。
上午:Transformer 直觉与 ESM-2 架构 (3h)
1.从 NLP 到 Protein NLP:
o讲解 Attention Map 如何对应蛋白质的接触图(Contact Map)。oMasked Language Modeling (MLM):为什么“完形填空”能学会蛋白质进化规律?
2.Embedding 的数学意义:
演示 Embedding 向量(如 1280 维)如何包含二级结构和功能信息。
下午:云端提取 Embeddings 实战 (3h)
1.Hugging Face transformers 库实战:
o代码实现:在 Colab 中加载 facebook/esm2_t33_650M_UR50D
o批量推理:将第一天清洗的 FASTA 序列转化为 Tensor 矩阵并保存为 .pt 文件。
2.潜空间(Latent Space)可视化:
o使用 UMAP 库对高维 Embedding 进行降维。
o案例分析:在 2D 平面上展示抗菌肽与非抗菌肽的聚类分离情况。
参考文献 (Confirmed):
·[ESM-2] Lin, Z., et al. (2023). “Evolutionary-scale prediction of atomic-level protein structure with a language model.” Science, 379(6637), 1123-1130.
·[可视化] McInnes, L., et al. (2018). “UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction.” arXiv:1802.03426.
生成式扩散模型部署 (Deployment of Generative Diffusion Models)
核心目标:复现论文核心。利用开源的 Diffusion 代码框架,在 Colab 上进行微调或推理。 (注:如果特定的 AMP-Diffusion 代码未开源,我们将使用通用的 Latent Diffusion 框架适配)
上午:扩散模型 (Diffusion) 原理精讲 (3h)
1.生成模型的代际演变:GAN vs VAE vs Diffusion。
2.去噪扩散概率模型 (DDPM):
o直观理解:从“高斯噪声”中雕刻出“蛋白质序列”。
条件引导 (Conditioning):如何告诉模型“给我生成一个带+4电荷的肽”?(Classifier-Free Guidance 原理)。
下午:AMP-Diffusion 云端复现 (3h)
1.加载预训练模型:
o从 GitHub 克隆代码仓库(模拟论文官方 Repo 或使用类似的 ProtDiff / Diffusers 框架)。
o加载 Checkpoint 权重。
2.条件生成实验:
o实验组 A:设定条件为“High Activity” + “Low Toxicity”。
o实验组 B:无条件随机生成。
o采样 (Sampling):执行 50-100 步的去噪过程(DDIM Sampler),生成 1000 条候选序列。
3.结果解码:将生成的 Latent Vector 映射回氨基酸序列文本。
��参考文献 (Confirmed & Supporting):
·[核心对标] Torres, M.D.T., et al. (2025). “Generative latent diffusion language modeling yields anti-infective synthetic peptides.” Cell Biomaterials. (注:以此为项目蓝本)
·[扩散模型基础] Ho, J., et al. (2020). “Denoising Diffusion Probabilistic Models.” NeurIPS.
·[蛋白质生成] Trippe, B. L., et al. (2023). “Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem.” ICLR.
高通量计算筛选 (In Silico High-Throughput Screening)
核心目标:构建“数字漏斗”,利用现有的预测工具 API 或 Python 包筛选生成序列。
上午:活性与毒性预测 (3h)
1.部署预测器:
o活性:在 Colab 中安装 AMP-Scanner-V2 (Keras/TensorFlow 版本)。
o毒性:使用 ToxinPred 的本地 Python 实现或批量调用 API。
2.批量评分脚本:
o编写 Pandas 脚本,对第三天生成的 1000 条序列进行打分。
设定阈值:保留 Probability > 0.8 的序列。

下午:多目标优化与排序 (3h)
1.理化性质复核:
o利用 modlAMP 库计算生成序列的疏水力矩。剔除那些结构上无法形成两亲性螺旋的“伪高分”序列。
2.新颖性控制 (Novelty Check):
o在 Colab 中运行本地 BLAST (NCBI-BLAST+)。
o策略:剔除与训练集相似度 > 60% 的序列,确保发现的是全新骨架。
3.最终榜单 (Leaderboard):
o输出 Top 20 候选序列及其六维雷达图(活性、毒性、溶血性、电荷、疏水性、新颖性)。
参考文献 (Confirmed):
·[AMP-Scanner] Veltri, D., et al. (2018). “Deep learning improves antimicrobial peptide recognition.” Bioinformatics, 34(16), 2740-2747.
·[多肽设计库] Müller, A. T., et al. (2017). “modlAMP: Python for antimicrobial peptide design.” Bioinformatics, 33(17), 2753-2755.
·[毒性预测] Gupta, S., et al. (2013). “In silico approach for predicting toxicity of peptides and proteins.” PLoS ONE, 8(9), e73957.
结构验证与 AlphaFold 实战 (Structural Validation via ColabFold)
核心目标:利用 AlphaFold 进行最终的物理验证,并产出科研级别的可视化图表。
上午:ColabFold 结构预测 (3h)
1.ColabFold 介绍:
o为什么 ColabFold 比原始 AlphaFold2 快 100 倍?(MMseqs2 加速原理)。2.云端折叠实战:
o将 Top 20 序列输入 ColabFold Notebook。
o关键指标解读:pLDDT(局部置信度)和 PAE(预测对齐误差)。
o注:对于 20-30 AA 的短肽,重点看是否形成了预期的 Alpha-Helix 结构。
下午:PyMOL 可视化与总结 (3h)
1.PyMOL 脚本作图:
o(本地或云端) 渲染表面电荷图 (Electrostatic Surface)。
o验证标准:是否呈现完美的“红蓝分离”(疏水面与带电面分离),这是破坏细菌膜的关键结构特征。
2.课程结业项目:
o每位学员提交一份 .csv 文件(含 Top 序列)和一张 .png 结构图。
o讨论:如何将这套流程迁移到其他任务(如食品风味肽、穿膜肽设计)。
参考文献 (Confirmed):
·[ColabFold] Mirdita, M., et al. (2022). “ColabFold: making protein folding accessible to all.” Nature Methods, 19(6), 679-682.
·[AlphaFold] Jumper, J., et al. (2021). “Highly accurate protein structure prediction with AlphaFold.” Nature, 596(7873), 583-589.
·[PyMOL] Schrodinger, LLC. (2015). “The PyMOL Molecular Graphics System, Version 1.8.”


上下滑动查看更多


第一天
启程·洞见药物设计的微观世界 (Vision & Foundation)
上午:理论基石——计算药物设计的思想与方法
·模块一:药物发现的源头——为何蛋白质三维结构至关重要?
o经典方法:同源建模(Homology Modeling)的逻辑与应用
o前沿探索:从头建模(de novo Modeling)的挑战与突破
·模块二:锁定靶心——蛋白质活性位点的识别与意义
o探索药物与靶点的“钥匙-锁”关系
·模块三:分子之舞——优秀药物分子的关键结构特征
o解构小分子药物的构效关系奥秘
·模块四:CADD核心武器库概览
o分子对接(Molecular Docking):预测分子间的“亲密接触”
o虚拟筛选(Virtual Screening):在百万分子中“大海捞针”
o分子动力学模拟(Molecular Dynamics):观测分子的动态行为
o其他前沿计算方法简介
下午:实战预备——可视化工具与数据解读
·模块五:解锁生命蓝图——蛋白质结构数据库(PDB)深度解析
o高效检索:精准定位你的目标蛋白
o信息解读:全面掌握PDB页面信息与生物学含义
o数据获取:标准数据的下载与管理
o破译密码:PDB文件格式的结构化解读
·模块六:PyMOL大师课——从入门到精通的分子可视化
o初识PyMOL:核心功能与界面导览
o基础操作:旋转、缩放、选择与着色
o高级渲染:绘制精美的蛋白/小分子表面图与静电势分布图
o成果展示:精准绘制相互作用图并制作引人注目的科学动画
第二天:构建·从序列到三维结构的创生 (From Sequence to Structure)
上午:同源建模专题——预测未知的蛋白质结构
·模块一:同源建模的原理与应用场景
o何时以及为何选择同源建模?
o揭秘同源建模背后的核心算法
·模块二:Swiss-Model实战工坊
o第一步:通过BLAST等工具寻找最佳同源模板
o第二步:序列比对的艺术与技巧
o第三步:精准选择决定模型质量的蛋白模板
o第四步:自动化与手动搭建蛋白三维模型
o第五步:模型质量的生命线——拉曼图(Ramachandran Plot)分析与解读
o第六步:模型的迭代优化与精修
·案例演练:以新冠病毒Spike蛋白序列为例,从零开始构建高质量三维结构模型,并进行全流程质量评估。
下午:小分子设计与数据库探索
·模块三:ChemDraw化学绘图实战
o从零开始,构建任意复杂的小分子结构
o一键计算:快速获取分子量、clogP等关键理化性质
o挑战练习:灵活构建大环分子、氨基酸、DNA/RNA片段等特殊结构
·模块四:探索海量的小分子化合物库
o主流数据库巡礼:DrugBank, ZINC, ChEMBL的特色与高效使用技巧
o特色资源探索:天然产物与中药成分数据库的挖掘与应用
第三天:对接·分子间的识别与结合 (The Docking Dance)
上午:分子对接的理论核心
·模块一:解密分子对接的黑箱
o核心原理:计算机如何模拟分子的结合过程?
o对接类型:刚性、半柔性与柔性对接的分类与选择
o评价标准:深入理解不同打分函数(Scoring Function)的物理意义
·模块二:常规分子对接流程实战
o配体准备:药物小分子的能量最小化与构象优化
o受体准备:蛋白靶点的预处理(加氢、去水、修复残基)
o定义靶点:受体结合口袋(Grid Box)的精确计算
o执行对接:运行高效的半柔性对接任务
下午:对接结果的深度分析与验证
·模块三:结果评估的多维视角
o黄金标准:与晶体结构(co-crystal structure)构象进行对比验证
o能量视角:从结合能与打分函数值评价结合强度
o聚类分析:洞察配体在口袋中的优势结合模式
o决策时刻:如何科学选择最优的结合构象?
·模块四:探索不同的对接策略与软件实现
第四天:进阶·柔性对接与虚拟筛选 (Advanced Docking & Screening)
上午:柔性对接——更真实的分子识别模拟
·模块一:柔性对接的核心流程
o配体的精细化准备与构象生成
o受体准备的特殊考量
o关键一步:定义需要柔性处理的氨基酸残基
o重新定义格点:适应受体柔性的计算盒子
o执行计算并进行结果分析与评估
·模块二:策略选择——半柔性 vs. 柔性对接
o深入比较两种方法的优缺点与适用场景
·模块三:柔性对接的替代实现方案
下午:高通量虚拟筛选——从海量分子中发现先导化合物
·模块四:药物发现的加速器
o小分子文件的“语言”:SDF, MOL2等格式的理解
o瑞士军刀OpenBabel:精通小分子格式的批量转化与处理
·模块五:基于对接的虚拟筛选实战
o流程构建:设计一套完整、高效的虚拟筛选工作流
o准备阶段:靶点蛋白的选择与百万级化合物库的获取
o执行阶段:自动化运行高通量对接任务
o结果分析:如何从海量结果中筛选出高潜力的“Hits”分子(Hit Identification)?
第五天:拓展·探索非经典分子相互作用 (Expanding the Horizon)
上午:特殊分子体系的对接挑战
·模块一:小分子-小分子对接
o超越蛋白靶点:探索小分子间的相互作用(例如:药物-糖类)
o独特的结构预处理与对接策略
o案例分析:以糖-小分子对接为例,解读结果并展示
·模块二:蛋白-核酸对接
o探索基因调控与药物干预的奥秘
·模块三:蛋白-蛋白对接(PPI)
otackling a new frontier in drug discovery
下午:QM/MM计算——融合量子化学与经典力学
·模块四:量子化学(QM)基础入门
o理论核心:薛定谔方程的启示
o方法概览:从半经验、HF到后HF方法
o中流砥柱:密度泛函理论(DFT)简介
·模块五:Gaussian计算入门示例
o从分子构建到单点能计算
o结构优化与振动分析
o探索化学反应:过渡态搜索与势能面扫描
·模块六:QM/MM在生物体系中的应用实例分析
o看QM/MM如何精确模拟酶催化反应等复杂过程
第六、七天:升华·分子动力学模拟与自由能计算 (Dynamics & Energetics)
第六天:驾驭Linux与GROMACS——开启MD模拟之旅
·上午:Linux系统基础
o迈入高性能计算的门槛:Linux系统简介
o效率倍增:常用核心命令实操
o软件安装:以GROMACS为例,掌握Linux环境下的程序部署
·下午:MD实战一:溶剂化环境中蛋白质的动态行为
o目标:全面掌握并亲手操作分子动力学模拟的完整标准流程(力场选择、构建体系、能量最小化、平衡、生产模拟)。
第七天:MD高级应用——配体结合与能量计算
·上午:MD实战二:蛋白质-配体复合物的动力学模拟
o挑战:学习处理非标准残基(如药物分子)的力场参数化与拓扑文件生成。
·下午:从模拟到定量——MD轨迹分析与自由能计算
o分析模块:掌握RMSD, RMSF, Radius of Gyration等常用分析方法
o定量模块:初步接触蛋白-配体结合自由能的计算方法(如MMPBSA/GBSA),定量评估结合强度
。
课程介绍:
本培训是系统的CADD课程培训,主要包含五种主要技术:Pymol画图、分子对接、虚拟筛选、QSAR建模、分子动力学模拟以及掌握这五种技术需要的辅助技术,如蛋白和小分子数据库的介绍和使用、蛋白和小分子的结构处理及性质分析、linux使用、openbabel使用等。
课程目标:
本课程能够让学员获得系统的CADD知识体系,独立完成科研工作中大部分CADD使用场景,具备自学特殊的高阶CADD方法的能力。


上下滑动查看更多


第一天
环境搭建与深度学习基本知识讲解
1.AIDD概述:从CADD到AIDD
2.软件安装与环境搭建
(1)anaconda
(2)vscode
(3)环境变量的配置
(4)切换pip和conda镜像源
(5)虚拟环境的创建
3.RDKIT工具包的使用
(1)基于RDKit的分子读写
(2)基于RDKit的分子绘制
(3)基于RDKit的分子指纹与分子描述符
(4)基于RDKit的化合物相似性与子结构
4.药物综合数据库的获取方法
(1)基于requests的基本爬虫操作
(2)小分子数据库PubChem数据获取(pubchempy / requests)
(3)蛋白质数据库PDB、UniProt数据获取
5.深度学习辅助药物设计
(1)神经网络基本概念与sklearn工具包介绍
(2)图神经网络与消息传递机制基本知识
(3)Transformer模型基本知识:分词、位置编码、注意力机制、编码器、解码器、预训练–微调框架、huggingface 生态介绍
(4)模型的评估与验证:准确率、精确率、召回率、F1分数、ROC曲线、AUC计算,平均绝对误差、均方差、R2分数、可释方差分数,交叉验证等
第二天
顶刊复现专题1——分子与生化反应的表示学习与性质预测助力药物发现
培训背景:在人工智能辅助药物发现(AIDD)中,分子与生化反应的表示学习与性质预测是整个研究流程的基石。分子的结构决定其功能,如何将复杂的分子结构和生化反应过程有效地表示为计算模型能够理解的形式,是实现高效预测和优化的前提。通过构建合理的分子表示(如图神经网络、SMILES编码、指纹等),我们可以让AI模型捕捉关键的化学特征,进而用于预测分子的物理化学性质、生物活性、毒性等,为后续的虚拟筛选、分子生成与反应设计提供可靠基础。因此,本专题不仅奠定了AIDD中建模与预测能力的核心能力框架,也为整个药物发现过程中的智能决策打下了坚实基础。
培训内容1:
Nature Machine Intelligence|基于注意力的神经网络在化学反应空间映射中的应用《Mapping the space of chemical reactions using attention-based neural networks》
1.数据集
1.1.Pistachio数据集:包含260万化学反应,来自专利数据,涵盖792个反应类别。数据经过去重和有效性过滤(使用RDKit)。
1.2.USPTO 1k TPL数据集:基于USPTO专利数据,包含44.5万反应,通过原子映射和模板提取生成1,000个反应模板类别。
1.3.Schneider 50k数据集:公开数据集,包含5万反应,50个类别,用于与传统指纹方法对比。
2.模型。研究对比了两种Transformer架构:
2.1.BERT分类器:基于编码器的模型,通过掩码语言建模预训练后,在分类任务上微调,使用[CLS]标记的嵌入作为反应指纹(rxnfp)。
2.2.Seq2Seq模型:编码器–解码器结构,将分类任务分解为超类、类别和具体反应的层级预测。两者均采用简化版BERT(隐藏层256维),输入为未标注的SMILES序列,无需反应物–试剂区分或原子映射。
3.训练。模型训练分为两步:
3.1.预训练:BERT通过掩码SMILES令牌预测任务进行自监督学习,学习反应通用表示。
3.2.微调:在分类任务上优化模型,使用交叉熵损失,学习率2×10⁻⁵,序列长度512。评估采用混淆熵(CEN)和马修斯相关系数(MCC)以处理数据不平衡。
培训内容2:
TOP期刊|基于深度学习的生化反应产量预测《Prediction of chemical reaction yields using deep learning》
1.数据。研究使用了三类数据:
1.1.Buchwald-Hartwig HTE数据集:包含3955个Pd催化C-N偶联反应,涵盖15种卤化物、4种配体、3种碱和23种添加剂组合,产率通过统一实验测量,数据质量高。
1.2.Suzuki-Miyaura HTE数据集:包含5760个反应,涉及15对亲电/亲核试剂、12种配体、8种碱和4种溶剂的组合,产率分布均匀。
1.3.USPTO专利数据集:从公开专利中提取,包含不同规模(克级与亚克级)的反应产率,数据噪声大且分布不一致,需通过邻近反应产率平滑处理以提升模型表现。
2.模型。核心模型基于预训练的rxnfp(反应指纹)BERT架构,新增回归层构成Yield-BERT。输入为标准化反应SMILES,通过自注意力机制捕捉反应中心及关键试剂的上下文信息。模型无需手工特征(如DFT计算描述符),直接端到端预测产率。实验表明,其性能优于传统方法(如随机森林和分子指纹拼接),尤其在HTE数据上接近化学描述符的预测水平,且参数鲁棒性高(超参数调整影响小)。
3.训练。训练分为两步:
3.1.预训练:BERT通过掩码语言任务学习SMILES的通用表示。
3.2.微调:采用简单Transformers库和PyTorch框架,以MSE损失优化回归层,学习率(2×10⁻⁵)和dropout率(0.1–0.8)为主要调参对象。HTE数据采用随机/时间划分验证,USPTO数据通过邻近反应产率平滑缓解噪声影响。小样本实验(5%训练数据)显示模型能快速筛选高产反应,指导合成优化。
培训内容3:
TOP期刊|基于T5Chem模型的生化反应表示学习与性质预测: 《Unified Deep Learning Model for Multitask Reaction Predictions with Explanation》
1.数据来源和处理。通过自监督预训练与PubChem分子数据集进行训练,以实现对四种不同类型的化学反应预测任务的优异性能。模型处理包括反应类型分类、正向反应预测、单步逆合成和反应产率预测。
2.模型架构和原理。T5Chem模型是基于自然语言处理中的“Text-to-Text Transfer Transformer”(T5)框架开发的统一深度学习模型,该模型通过适应T5框架来处理多种化学反应预测任务。T5Chem模型包含编码器–解码器结构,并根据任务类型引入了任务特定的提示和不同的输出层,如分子生成头、分类头和回归头,以处理序列到序列的任务、反应类型分类和产品产率预测。
3.训练过程和细节。
3.1.T5Chem模型首先在PubChem的97 million分子上进行自监督预训练,使用BERT类似的“masked language modeling”目标。
3.2.在预训练阶段,源序列中的tokens被随机掩蔽,模型的目标是预测被掩蔽的正确的tokens。
3.3.预训练完成后,模型在下游的监督任务中进行微调,使用不同的任务特定提示和输出层。
3.4.模型在测试阶段通过生成分子token by token的方式进行预测,直到生成“句子结束标记”或达到最大预测长度。
通过培训可以掌握的内容:
1.分子与化学反应的表示方法。学习如何将分子和化学反应编码为机器可处理的格式,如SMILES(Simplified Molecular-Input Line-Entry System)和反应SMILES。理解分子指纹(如Morgan指纹)和反应指纹(如rxnfp)的构建方式,以及它们在化学信息学中的应用。掌握Transformer架构(如BERT)如何用于化学反应的特征提取,并生成具有化学意义的向量表示。
2.深度学习在化学反应预测中的应用。了解如何利用序列到序列(Seq2Seq)模型和BERT进行化学反应分类(如反应类型识别)和产率预测。学习如何通过自注意力机制分析化学反应的关键部分(如反应中心、试剂影响),提高模型的可解释性。掌握如何利用预训练+微调策略,使模型在少量标注数据下仍能取得良好性能。
3.数据驱动的化学研究范式。认识不同数据来源(如高通量实验HTE、专利数据USPTO)的特点及其对模型训练的影响。学习如何处理数据噪声,并通过数据平滑、邻近分析等方法优化模型表现。了解小样本学习在化学中的应用,例如如何用5%-10%的训练数据筛选高产率反应,指导实验优化。
第三天
顶刊复现专题2——蛋白质的表示学习与性质预测助力药物发现
培训背景:在AIDD中,蛋白质是药物作用的主要靶标,其结构与功能的复杂性决定了药物设计的成败。蛋白质的表示学习与性质预测是理解分子–靶点相互作用、发现候选药物的重要环节。蛋白质,尤其是酶,作为药物的主要作用靶点,其功能、结构与动力学性质直接影响药物的设计与效果。本专题通过两篇前沿研究工作展开讲解:*《Enzyme function prediction using contrastive learning》展示了如何利用对比学习从蛋白质序列中提取高质量的功能表征,实现对酶功能的精确预测;《CatPred》*则提出了一个整合性深度学习框架,用于体外酶动力学参数(如Km、kcat等)的预测,这对于建立药效模型与优化先导化合物至关重要。这些方法显著提升了蛋白质建模的准确性与泛化能力,为AI驱动的靶点发现、机制理解及候选药物筛选提供了强有力的支持。
培训内容1:
Nature Communication|体外酶动力学参数深度学习的综合框架《CatPred: a comprehensive framework for deep learning in vitro enzyme kinetic parameters》
CatPred 提出了一种全面的深度学习框架,用于预测体外酶动力学参数(kcat、Km、Ki),以解决实验测定成本高、数据稀疏和泛化能力差的问题。该方法不仅提供了准确的预测,还引入了对预测不确定性的量化,支持对训练集外(out-of-distribution)酶序列的稳健预测。此外,作者还构建了新的标准化数据集(CatPred-DB),并对多种酶表示方法进行了系统比较。
1.数据:CatPred 使用的数据集来自 BRENDA 和 SABIO-RK 数据库,作者构建了 CatPred-DB,包括:23197 条 kcat,41174 条 Km和11929 条 Ki 数据,每条记录都包含酶的氨基酸序列、AlphaFold 或 ESMFold 预测的结构、底物的 SMILES 表达式。数据经过清洗和标准化处理,去除缺失值和重复值,并对参数取对数转换以符合正态分布。
2.模型:CatPred 采用模块化设计,酶和底物分别通过不同的神经网络模块进行表征学习,并采用 概率回归 输出(高斯分布形式的均值和方差),允许进行 不确定性估计(aleatoric + epistemic)。
3.训练
3.1.所有模型采用负对数似然损失函数(NLL)训练,以同时预测参数均值和不确定性。
3.2.使用训练–验证–测试三分法(80%-10%-10%),并设立“训练集外”的测试子集用于泛化能力评估。
3.3.为了评估不确定性,CatPred 使用 10个模型的集成,通过不同初始参数训练,以此量化 epistemic uncertainty。
3.4.模型训练时考虑了不同相似性(序列identity<99%、80%、60%、40%)的测试集,体现其鲁棒性。
培训内容2:
Science|基于对比学习的蛋白质分类属性预测《Enzyme function prediction using contrastive learning》
1.数据来源和处理: CLEAN模型的训练基于UniProt数据库中的高质量数据,该数据库收录了约1.9亿个蛋白质序列。CLEAN模型以氨基酸序列作为输入,输出按可能性排序的酶功能列表(以EC编号为例)。为了验证CLEAN的准确性和鲁棒性,作者进行了广泛的in silico实验,并将CLEAN应用于内部收集的未表征的卤酶数据库(共36个)进行EC编号注释,随后通过案例研究进行体外实验验证。
2.模型架构和原理: CLEAN模型采用了对比学习框架,目标是学习一个酶的嵌入空间,其中欧几里得距离反映了功能相似性。嵌入是指蛋白质序列的数值表示,它由机器可读,同时保留了酶携带的重要特征和信息。在CLEAN的任务中,具有相同EC编号的氨基酸序列具有较小的欧几里得距离,而具有不同EC编号的序列则具有较大的距离。
3.训练过程和细节:
3.1.在训练过程中,CLEAN模型使用对比损失函数进行监督训练,通过优先选择与锚点(anchor)嵌入具有小欧几里得距离的负序列,以提高训练效率。
3.2.模型使用语言模型ESM1b获得的蛋白质表示作为前馈神经网络的输入,输出层产生细化的、功能感知的输入蛋白质嵌入。
3.3.预测时,通过计算查询序列与所有EC编号聚类中心之间的成对距离来预测输入蛋白质的EC编号。
3.4.CLEAN还开发了两种方法来从输出排名中预测自信的EC编号:一种是贪婪方法,另一种是基于P值的方法。
通过培训可以掌握的内容:
1. 蛋白质表示学习的基本方法:理解蛋白质序列、结构信息如何被编码为适用于深度学习模型的向量表示,包括基于序列的预训练模型(如ESM、ProtBERT)和结构感知模型的原理与应用。
2. 对比学习在生物功能预测中的应用:深入学习对比学习策略,掌握如何通过正负样本构建来提升蛋白质功能分类模型的判别能力。
3. 酶动力学参数预测建模框架:学员将理解如何结合序列、结构及辅助特征,利用深度学习模型预测关键的酶学参数(如Km、kcat),并掌握模型架构设计与性能评估的思路。
4. 评估与可解释性分析方法:学习如何分析模型预测结果,评估性能指标,并探索特征重要性等可解释性技术,帮助理解模型的决策依据。
5. 应用于真实药物研发场景的思维框架:建立从蛋白质建模到下游任务(如药物筛选、作用机制分析)的系统性理解,增强将AI方法应用于实际生物医药问题的能力。
第四天
顶刊复现专题3——基于深度学习的分子生成助力药物发现
培训背景:分子生成是化学、生物学和材料科学等领域的关键技术,对于新药开发、新材料设计和化学反应预测具有重要意义。传统的分子生成方法依赖于专家知识和试错实验,耗时且成本高昂。随着人工智能技术的发展,特别是自然语言处理和扩散模型在分子生成中的应用,我们现在能够利用计算模型来加速这一过程。本课程将介绍从NLP到扩散模型的设计模式,这些模型能够理解和生成分子结构,从而提高分子设计的效率和准确性。通过本课程的学习,参与者将能够掌握分子生成的最新技术和方法,以及如何将这些技术应用于实际问题。
培训内容1:
Nature Communication|基于端到端的图生成框架的分子生成:《Retrosynthesis prediction using an end-to-end graph generative architecture for molecular graph editing》
1.数据来源和处理:Graph2Edits模型使用了公开可用的基准数据集USPTO-50k,包含50016个反应,这些反应被正确地原子映射并分类为10种不同的反应类型。数据集被分为40k、5k、5k的反应用于训练、验证和测试集。
2.模型架构和原理:Graph2Edits模型是一个端到端的图生成架构,基于图神经网络(GNN)预测产品图的编辑序列,并根据预测的编辑序列顺序生成中间体和最终反应物。该模型将半模板方法的两阶段过程(识别反应中心和完成合成子)合并为一锅学习,提高了在复杂反应中的适用性,并使预测结果更易于解释。模型的核心是图编码器和自回归模型,用于生成编辑序列,并应用这些编辑来推断中间体和反应物。
3.训练过程和细节:
3.1.Graph2Edits模型使用有向消息传递神经网络(D-MPNN)作为图编码器,以获取原子表示和全局图特征,并预测原子/键编辑和终止符号。
3.2.模型训练使用教师强制策略,即使用真实的编辑序列作为模型输入。在每个编辑步骤中,模型会计算所有可能的编辑的概率,并选择最高分的k个编辑,将这些编辑应用于输入图以获得k个中间体。
3.3.在生成过程中,如果达到最大步骤数或图表示指示终止,则生成分支将停止。
3.4.最终,根据可能性对前k个编辑序列和图进行排名,收集为最终预测结果。
培训内容2
Nature Computational Science|基于等变扩散模型的分子生成网络《Structure-based drug design with equivariant diffusion models》
1.简单介绍。这篇文献提出了一种基于结构的药物设计方法(SBDD),利用SE(3)-等变扩散模型(DiffSBDD)生成与蛋白质结合口条件匹配的新颖小分子配体。该方法通过将SBDD问题建模为三维条件生成任务,能够一次性生成所有原子位置,克服了传统自回归方法因顺序生成而丢失全局上下文的局限性。DiffSBDD不仅支持从头分子设计,还能通过属性优化、负向设计和分子局部修饰(inpainting)等多种任务灵活应用。
2.数据总结。该研究使用了CrossDocked和Binding MOAD两个数据集进行训练和评估。
2.1.CrossDocked数据集包含40,344个训练蛋白–配体对和130个测试对,验证集规模为246个,确保不同集合中的蛋白质来自不同的酶分类主类以避免过拟合。
2.2.Binding MOAD数据集经过筛选后用于测试,分析限于所有方法均能生成样本的78个CrossDocked和119个Binding MOAD目标。此外,数据集处理涉及移除损坏条目,并通过Zenodo公开提供处理后的数据和采样分子,确保研究可重复性。
3.模型总结。DiffSBDD是一个SE(3)-等变扩散模型,以蛋白质结合口为条件生成三维分子结构,采用3D图表示(原子坐标和类型),避免了传统方法中从密度图回推分子结构的复杂后处理。模型设计尊重三维空间的旋转和平
通过培训可以掌握的内容:
1.自然语言处理(NLP)在分子生成中的应用:掌握如何使用NLP技术来理解和生成分子结构。学习如何将自然语言描述转换为分子结构(SMILES字符串)。
2.扩散模型在分子生成中的应用:理解扩散模型的基本原理及其在分子生成中的优势。学习如何使用扩散模型来优化分子生成过程。
3.数据预处理和特征工程:学习如何处理和准备用于训练分子生成模型的数据集。掌握如何从原始数据中提取有用的特征以提高模型性能。
4.模型架构和原理:深入理解MolT5,TGM-DLM和GraphEdits模型的架构和工作原理。学习如何设计和实现这些模型以处理复杂的分子生成任务。
5.训练过程和细节:掌握模型训练的全过程,包括预训练和微调。学习如何调整模型参数和训练策略以优化性能。
6.评估和验证:学习如何使用各种指标(如BLEU分数、Tanimoto相似性等)来评估生成的分子。掌握如何验证模型生成的分子的有效性和准确性。
7.模型解释和可视化:学习如何解释模型的预测结果,以及如何使用可视化工具来理解分子生成过程。
8.最新研究进展和技术趋势:了解分子生成领域的最新研究进展和技术趋势。学习如何将最新的研究成果应用于实际工作。
第五天
顶刊复现专题4: 结合分子动力学的蛋白质–配体复合物相互作用动态预测
培训背景:蛋白质–配体相互作用的预测是现代药物发现和生物工程领域的核心任务之一,其重要性不言而喻。在药物开发过程中,准确预测蛋白质与小分子配体的结合位点、三维结构以及亲和力,不仅能够揭示分子间相互作用的机制,还能显著加速候选药物的筛选与优化,降低研发成本和时间。传统实验方法如X射线晶体学和核磁共振虽然精确,但耗时长、成本高,且难以应对大规模筛选需求。而随着深度学习和人工智能技术的快速发展,计算方法在蛋白质–配体预测中展现出巨大潜力。
研究内容1:
Nature Communication|交互作用感知的蛋白质–配体对接和亲和力预测模型《Interformer: an interaction-aware model for protein-ligand docking and affinity prediction》
1.简要介绍:本研究提出了一种名为Interformer的基于Graph-Transformer架构的统一模型,用于蛋白–配体对接和亲和力预测。针对现有深度学习模型忽略蛋白与配体原子间非共价相互作用建模的不足,Interformer引入了交互感知混合密度网络(MDN)来明确捕捉氢键和疏水相互作用,并结合负采样策略和伪Huber损失函数,通过对比学习优化相互作用分布,提升对接姿势的准确性和亲和力预测的鲁棒性。
2.数据集:研究使用了PDBBind时间分割测试集(333个样本)评估对接准确性,Posebusters基准测试验证物理合理性,以及内部真实世界数据集测试泛化能力。训练数据来源于PDBBind晶体结构数据库。
3.模型:Interformer基于Graph-Transformer架构,包括:(1) 图表示模块,将原子作为节点、邻近关系作为边;(2) 掩码自注意力(MSA)机制,通过Intra-Blocks和Inter-Blocks分别捕捉配体/蛋白内部及两者间的相互作用;(3) 交互感知MDN,融合四种高斯分布模拟常规力、疏水作用和氢键;(4) 边缘输出层整合节点和边特征预测能量;(5) 姿势评分和亲和力模块基于虚拟节点预测正确姿势和实验亲和力值。
4.训练细节:训练分两阶段:首先基于晶体结构训练能量模型生成负样本,随后联合正负样本训练姿势评分和亲和力模型。采用负对数似然损失优化MDN,二元交叉熵损失优化姿势评分,伪Huber损失(σ=4)优化亲和力预测(单位IC50、Kd、KI,经负对数归一化)。蒙特卡洛采样生成候选姿势,
研究内容2:
Nature Communication|分子动力学驱动的蛋白质–配体复合物结构动态预测《DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model》
1.简单介绍:本研究提出了一种名为DynamicBind的深度学习方法,用于预测配体特异性的蛋白–配体复合物结构。传统分子对接方法通常将蛋白视为刚性或仅部分柔性,难以处理蛋白的大尺度构象变化,而分子动力学模拟虽然能捕捉动态构象,但计算成本高昂。DynamicBind通过等变几何扩散网络构建平滑的能量景观,高效模拟蛋白从无配体(apo)状态到配体结合(holo)状态的构象转变,无需依赖holo结构或大量采样。
2.数据集:研究基于PDBbind2020数据库(19,443个蛋白–配体复合物晶体结构),按时间划分:2019年前的数据用于训练和验证,2019年的数据用于测试。额外构建了Major Drug Targets (MDT)测试集(599对),聚焦激酶、GPCR等主要药物靶点,要求AlphaFold预测结构与晶体结构的pocket RMSD>2Å,确保测试难度。训练中通过AlphaFold预测结构与晶体结构插值生成蛋白部分的样本。
3.模型:DynamicBind是一个基于图神经网络的等变生成模型,使用粗粒化表示(蛋白以Cα节点和侧链二面角表示,配体以重原子节点表示),输出包括蛋白和配体的平移、旋转、扭转角更新,以及结合亲和力和cLDDT置信度评分。模型通过学习从apo到holo的“morph-like”变换,优化能量景观,包含63.67百万参数。
4.训练细节:训练在8块Nvidia A100 80GB GPU上进行5天,输入为添加morph变换的蛋白decoy构象和加高斯噪声的配体构象,目标是去噪操作。损失函数包括八项(配体和蛋白的平移、旋转、扭转等),通过Kabsch算法对齐apo和holo结构,结合扩散噪声调整构象过渡。推理时迭代20次更新初始结构。
通过培训可以掌握的内容:
1.蛋白质–配体复合物结构预测:学员将学习如何利用深度学习方法(如NeuralPLexer)从蛋白序列和配体分子图预测复合物的三维结构,理解多尺度几何建模和扩散过程在捕捉原子级分辨率结构及构象变化中的作用,并掌握其在盲对接和柔性结合位点恢复中的应用。
2.对接姿势生成与优化:掌握基于Graph-Transformer架构和蒙特卡洛采样生成对接姿势的技术,学习如何通过姿势评分和对比学习(如伪Huber损失)优化姿势选择,提升对接准确性(如RMSD<2Å的成功率)。
3.亲和力预测的计算方法:学员将了解如何从对接姿势预测实验亲和力值(如IC50、Kd、KI),掌握基于虚拟节点和对比学习的姿势敏感性训练策略,以提高亲和力预测的鲁棒性和实际应用价值。
4.模型评估与基准测试:熟悉常用基准数据集(如PDBBind)和评价指标(如RMSD、lDDT-BS、TM-score)的使用,理解如何通过时间分割测试集和物理合理性检查评估模型的泛化能力和性能。
5.实际药物设计的应用:通过案例分析(如Interformer筛选出高亲和力小分子),学习如何将这些预测技术应用于酶工程和药物发现,加速候选分子的筛选和优化过程。


上下滑动查看更多


第一天
合成生物学基础概念与应用领域剖析
1.深度解读合成生物学精准定义,系统阐述其涵盖研究内容,沿着发展历程脉络回溯,展望未来趋势,全方位扫描应用领域。
2.通过详实案例分析,将合成生物学在医药、农业、工业等实际场景的应用具象化,让学员感知其变革力量。
生物元件功能精讲与标准化设计准则
1.聚焦生物元件,如启动子精准调控转录起始、终止子界定转录终点、RBS 驱动核糖体结合开启翻译。
2.结合 BioBrick 元件设计范式与应用实例,传授生物元件模块化设计黄金法则,助力学员掌握构建标准化生物模块的精髓。
第二天
基因线路逻辑架构搭建与实例演练
1.开启基因线路逻辑世界大门,详细拆解与、或、非基本逻辑门运作原理及设计技巧。
2.引入 iGEM 竞赛中经典逻辑门线路应用案例,手把手指导学员实践操作,使其能独立构思简单基因线路并洞悉其功能。
复合元件整合与电路深度设计
以前两天所学生物元件、基因线路知识为基石,引导学员梳理合成生物学核心玩法。亲手设计、搭建、分析经典电路,实现知识融会贯通,掌握复杂电路中各模块协同运作奥秘。
第三天
代谢途径精细构建与优化策略
1.深入讲解代谢途径搭建步骤,剖析定向进化、代谢工程等优化策略。
2.以青蒿素生物合成途径优化为典型案例,传授从理论到实践的代谢途径设计方法,助力学员提升生物合成效率。
基因组合成与编辑技术前沿
1.系统讲解基因组合成底层原理、多元方法,深度剖析 CRISPR/Cas9 等基因组编辑技术原理。
2.结合海量案例,呈现基因组合成与编辑技术在定制生物、精准医疗等前沿领域的实际应用。
第四天
底盘生物特性解析与应用
1.引入底盘生物概念,剖析常见底盘生物(如大肠杆菌、酵母菌等)特性。
2.讲解其在基因线路搭载、基因表达中的关键作用,结合实例探讨如何依据项目需求选择适配底盘生物,实现高效基因操作。
合成生物系统搭建与优化实践
1.全方位讲解合成生物系统定义、分类,通过案例剖析系统构建流程,传授优化策略。
2.组织实践操作,让学员亲身体验合成生物系统的构建与优化全过程,学会依据反馈调控系统参数。
第五天
合成生物学数学建模与性能分析
1.开启生物系统数学建模之旅,介绍常微分方程、逻辑模型等常用建模方法。
2.结合基因线路动力学模拟案例,指导学员运用数学模型精准分析生物系统性能,为系统设计与优化提供量化支撑。
合成生物学全景总结与未来瞻望
1.回溯课程所学知识,通过多领域案例分析,展现合成生物学应用广度与深度。
2.组织学员热烈讨论未来发展趋势、潜在影响,着重介绍伦理、生物安全与生物安保等关键议题,培养学员全面视角。
学习目标:通过理论与实践结合,掌握合成生物学基础、基因电路设计、代谢途径优化、基因编辑技术及数学建模,培养学员在合成生物领域的创新能力和系统思维,为未来研究与应用奠定基础。


上下滑动查看更多


第一天
A1 代谢物及代谢组学的发展与应用
(1) 代谢与生理过程;
(2) 代谢与疾病;
(3) 非靶向与靶向代谢组学;
(4) 空间代谢组学与质谱成像(MSI);
(5) 代谢组学与药物和生物标志物;
(6) 代谢流与机制研究。A2 代谢通路及代谢数据库
(1) 几种经典代谢通路简介;
(2) 三大常见代谢物库:HMDB 、METLIN 和 KEGG;
(3) 代谢组学原始数据库:Metabolomics Workbench 和 Metabolights. A3 参考资料推荐
A4 代谢组学实验流程简介
A5 色谱 、质谱硬件与原理解析
(1) 色谱分析原理与构造;
(2) 色谱仪和色谱柱的选择;
(3) 色谱的流动相:梯度洗脱法;
(4) 离子源、质量分析器与质量检测器解析;
(5) 质谱分析原理及动画演示;
(6) 色谱质谱联用技术(LC-MS);
第二天
B1 代谢物样本处理与抽提
(1) 各种组织、血液和体液等样本的提取流程与注意事项;
(2) 代谢物抽提流程与注意事项;
(3) 样本及代谢物的运输与保存问题;
B2 LC-MS 数据质控与搜库
(1) LC-MS 实验过程中 QC 和 Blank 样本的设置方法;
(2) LC-MS 上机过程的数据质控监测和分析;
(3) 代谢组学上游分析原理——基于 Compound Discoverer 与 Xcms 软件;
(4) Xcms 软件数据转换、提峰、峰对齐与搜库;
B3 R 软件基础
(1) R 和 Rstudio 的安装;
(2) Rstudio 的界面配置;
(3) R 中的基础运算和统计计算;
(4) R 中的包:包,函数与参数的使用;
(5) R 语言语法,数据类型与数据结构;
(6) R 基础画图;
B4 ggplot2
(1) ggplot2 简介
(2) ggplot2 的画图哲学;
(3) ggplot2 的配色系统;
(4) ggplot2 数据挖掘与作图实战;
第三天
机器学习
C1 有监督式机器学习在代谢组学数据处理中的应用
(1) 人工智能、机器学习、深度学习的关系;
(2) 回归算法:从线性回归、Logistic 回归与 Cox 回归讲起;
(3) PLS-DA 算法:PCA 降维后没有差异的数据还有救吗?
(4) VIP score 的意义及选择;
(5) 分类算法:决策树,随机森林和贝叶斯网络模型;
C2 一组代谢组学数据的分类算法实现的 R 演练
(1) 数据解读;
(2) 演练与操作;
C3 无监督式机器学习在代谢组学数据处理中的应用
(1) 大数据处理中的降维;
(2) PCA 分析作图;
(3) 三种常见的聚类分析:K-means、层次分析与 SOM
(4) 热图和 hcluster 图的 R 语言实现;
C4 一组代谢组学数据的降维与聚类分析的 R 演练
(1) 数据解析;
(2) 演练与操作;
第四天
D1 在线代谢组分析网页 Metaboanalyst 操作
(1) 用 R 将数据清洗成网页需要的格式;
(2) 独立组、配对组和多组的数据格式问题;
(3) Metaboanalyst 中的上游分析(原始数据峰提取、峰对齐与搜库)
(4) Metaboanalyst 的 pipeline 以及参数设置和注意事项;
(5) Metaboanalyst 的结果查看和导出;
(6) Metaboanalyst 的数据编辑;
(7) 全流程演练与操作。
D2 代谢组学数据清洗与 R 语言进阶
(1) 代谢组学中的 t、fold-change 和响应值;
(2) 数据清洗流程;
(3) R 语言 tidyverse;
(4) 数据预处理:数据过滤与数据标准化(样本的 Normalization 和代谢物的 Scaling);
(5) 代谢组学数据清洗演练;
第五天
E1 文献数据分析部分复现(1 篇)
(1) 文献深度解读;
(2) 实操:从原始数据下载到图片复现;
(3) 学员实操。
E2 机器学习与代谢组学顶刊解读(3 篇);
(1) Signal Transduction and Targeted Therapy 一篇有关饥饿对不同脑区代谢组学影响变
化的小鼠脑组织代谢图谱类的文献;(数据库型)
(2) Cell 一篇代谢组学孕妇全程血液代谢组学分析得出对孕周和孕产期预测的代谢标志物
的文献;(生物标志物型)
(3) Nature 一篇对胰腺癌患者肠道菌群的代谢组学分析找到可以提高化疗效果的代谢物的
文献。(机制研究型)


上下滑动查看更多


第一天
多组学测序技术及数据库
上午、理论讲解
1. 多组学测序技术
2. 介绍多组学数据库
3. 深度学习融合多组学模型及应用介绍
GPU 服务器上机实操
1.Linux 操作系统
1.1 常用的Linux 命令
1.2 Vim 编辑器
1.3 基因组数据文件管理, 修改文件权限
1.4 查看探索基因组区域
2.Python 语言基础
2.1.Python 包安装和环境搭建
2.2.常见的数据结构和数据类型
下午、深度学习实现多组学数据插补模型理论讲解Python代码解析及GPU服务器上机实操
1. 多组学融合通用框架模型CustOmics
2. 非监督深度学习癌细胞系合成数据增强模型MOSA (Multi-Omic Synthetic Augmentation)
第二天
深度学习识别基因变异及疾病亚型
上午、深度学习识别基因变异模型理论讲解Python代码解析及GPU服务器上机实操
1. 深度学习识别基因变异诊断阿尔茨海默病SWAT
2. 多阶段融合多组学表观遗传数据预测转录因子深度学习模型TRAPT
下午、深度学习识别疾病亚型模型Python代码解析及GPU服务器上机实操
1. 多组学识别癌症亚型生成对抗式深度学习模型Subtype-GAN
2. 多尺度可解释的多组学深度学习模型DeepOmix预测癌症生存期
3. 联邦深度学习多组学数据预测癌症演化DeepProg模型
第三天
深度学习识别疾病标志物
上午、深度学习模型识别疾病标志物Python代码解析及GPU服务器上机实操
1. 多组学特征排序识别COVID-19疾病标志物Deep IDA模型
2. 基于肠道微生物组预测肠道代谢物高可解释性神经编码器-解码器网络模型BioNED
下午、深度学习模型识别病理图像标志物Python代码解析及GPU服务器上机实操
1. 基于深度学习的集成方法从组织病理学图像预测胃腺癌分子亚型DEMoS
2. 基于深度学习的结直肠癌病理图像预后标志物挖掘DigiPathAI
第四天
深度学习融合单细胞多组学数据
上午、深度学习融合单细胞多组学模型Python代码解析及GPU服务器上机实操 1. 单细胞多组学聚类多模态深度学习模型scMDC
2. 基于深度学习的生成式模型融合单细胞多组学数据scMM (mixture-of-experts deep generative model)
下午、融合单细胞空间多组学深度学习模型Python代码解析及GPU服务器上机实操
1. 空间反卷积多尺度深度模型TACIT推断细胞类型及细胞状态
2. 深度学习模型从单细胞数据解析醣基化生物过程
第五天
深度学习融合多模态功能学习识别疾病通路、药物重定位
上午、深度学习模型融合多模态功能学习识别疾病通路Python代码解析及GPU服务 器上机实操
1. 基于Transformer 的深度学习模型整合多组学数据与癌症通路DeePathNet
2. 一种识别泛癌种Ras通路激活的深度学习方法NatDRAPl
下午、深度学习模型多组学整合药物重定位Python代码解析及GPU服务器上机实操
1. 基于核方法的深度学习框架实现多组学整合的药物重定位DeepDRK
2. 基于蛋白质相互作用网络嵌入细胞系以预测抗癌协同药物组合模型PRODeepSyn


上下滑动查看更多
01、人工智能构建虚拟细胞
本课程以虚拟细胞(Virtual Cell)构建为总体框架,系统拆解当前国际前沿细胞人工智能模型在多组学建模、机制推理、药物设计预测、疾病系统重构及数字孪生中的完整技术路径。课程以理论体系建立为核心,结合当前国际主流代表模型(如 MultiVI、scVI、DrugCell等)进行结构化解析,使学员能够从单点模型理解上升到整体建模逻辑理解。
02、AI蛋白质设计
本课程是一门面向生物医学领域学生和研究人员的高阶技术课程。课程旨在系统性地讲解AI在蛋白质设计领域的关键模型原理,并结合代码实践,最终让学员掌握一套完整的、可在本地部署的、从头(de novo)设计蛋白质的计算流程。
03、AI抗菌肽设计
本课程以“构建 AI 抗菌肽发现与设计流水线”为核心主题,面向生命科学、计算生物学、生物信息学及人工智能交叉领域,系统培养学员在 AI 驱动生物分子设计领域的理论理解能力与实践能力。课程围绕“数据 → 表征 → 生成 → 筛选 → 结构验证”的完整研发流程,构建可复现的计算抗菌肽设计体系。
04、CADD计算机辅助药物设计
本培训是系统的CADD课程培训,主要包含五种主要技术:Pymol画图、分子对接、虚拟筛选、QSAR建模、分子动力学模拟以及掌握这五种技术需要的辅助技术,如蛋白和小分子数据库的介绍和使用、蛋白和小分子的结构处理及性质分析、linux使用、openbabel使用等。
05、AIDD人工智能药物发现与设计
本课程让学员了解药物发现的前沿背景,学习人工智能领域的各类常见算法,熟悉工具包的安装与使用,掌握一定的算法编程能力,能够运用计算机方法研究药物相关问题。通过大量的案例讲解和实践操作,具备一定的AIDD模型构建和数据分析能力
06、合成生物学与基因电路设计
通过理论与实践结合,掌握合成生物学基础、基因电路设计、代谢途径优化、基因编辑技术及数学建模,培养学员在合成生物领域的创新能力和系统思维,为未来研究与应用奠定基础。
07、机器学习代谢组学
熟悉代谢组学和机器学习相关硬件和软件;熟悉代谢组学从样本处理到数据分析的全流程;能复现至少1篇CNS或子刊级别的代谢组学文章图片。
08、深度学习在多组学融合中的应用
课程通过基础入门+应用案例实操演练的方式,从初学及应用研究的角度出发,带大家实战演练多种深度学习模型(深度神经网络DNN、卷积神经网络CNN、循环神经网络RNN、自动编码器AE、图卷积神经网络GCN)在多组学融合分析中的各种应用
讲师介绍
01
人工智能构建虚拟细胞
生物学博士,深耕生命科研,转化医学及药物研发领域,先后任职于新加坡淡马锡生命科学院, 新加坡科学技术研究局等机构,系统追踪基因与细胞治疗, 抗体及ADC等药物研究领域,学术成果发表于Molecular cell, ASCO, ESMO 等国际会议及期刊,拥有丰富的产业实践经验。
02
AI蛋白质设计
授课老师均来自北京大学和清华大学,专注于蛋白设计和多肽药物靶点,具有丰富的代码开发经验和授课教学经验。在对应领域有过多篇中科院一区top期刊/CCF-A会议,如ICLR,ICML等主流会议的第一作者和审稿人经历。
03
AI抗菌肽设计
X老师,长期从事 AI辅助蛋白质设计、计算药物发现、多肽开发研究,在人工智能与生命科学交叉领域具有丰富的科研与教学经验,在 Nature 子刊,PNAS等国际期刊发表论文数篇
04
CADD计算机辅助药物设计
Z老师,多年Ai4science研究经验,研究方向为蛋白设计、药物发现与计算生物学。曾获多项国家级奖项,在计算机辅助药物设计,药物发现等领域具有丰富的理论知识和实践经验。
05
AIDD人工智能药物发现与设计
AIDD授课老师曹老师,有十余年的计算机算法研究和程序设计经验。研究方向涉及生物信息学,深度学习,药物合成路径设计,药物不良反应等。发明专利5项,参与国家重点科研项目4项,发表SCI高水平论文10篇,包括BMC Bioinformatics, Journal of Biomedical Informatics, International Journal of Molecular Sciences等知名期刊。
06
合成生物学与基因电路设计
两位授课老师均来自清华大学,干湿结合分别引领本课程的实验设计和建模分析,研究方向涉及植物生物学、合成生物学与生物信息学。在对应领域中科院一区有多篇产出,同时曾作为队长和评委多次参加过合成生物学(iGEM)顶级赛事,曾获得全球十佳项目(TOP10)和多个单项奖及提名。
07
机器学习代谢组学
主讲老师来自985高校,主要利用代谢组学、转录组学和分子生物学等技术研究神经内科慢性病的发病机制和生物标志物。擅长高效液相色谱-质谱联用(LC-MS)技术进行非靶向和靶向代谢组学从样本制备到数据分析的全流程研究,以及多组学大数据的生物信息学整合分析。5年内在J Clin Invest, EBioMedicine, Cell Death Dis, Cell Death Discov, Nanotoxicology等杂志发表SCI论文10篇。
08
深度学习在多组学融合中的应用
各讲老师刘老师,生物信息学博士,从事医学生物信息及人工智能研究15年,曾在新加坡基因组研究院及美国加州大学洛杉矶分校研究多组学数据在复杂疾病诊疗中的应用。研究领域涉及人工智能、自然语言处理、功能基因组学、宏基因组学、转录组学、miRNA 及靶基因网络分析,单细胞测序数据分析,基因调控网络时序分析,蛋白质互作网络分析,多组学联合分析等。主持省级自然科学基金等项目4项,开发过数个生物信息学工具,发表SCI论文20余篇,其中人工智能算法文章10余篇,编著医学数据分析实用教材一部。
JOIN US
授课时间
北京时间
.人工智能构建虚拟细胞
2026.05.18晚上授课(19:00-22:00)
2026.05.19晚上授课(19:00-22:00)
2026.05.20晚上授课(19:00-22:00)
2026.05.21晚上授课(19:00-22:00)
AI蛋白质设计
AI抗菌肽设计
CADD计算机辅助药物设计
2026.05.24全天(9点-11点–2点-5点)
2026.05.25晚上授课(19:00-22:00)
2026.05.26晚上授课(19:00-22:00)
2026.05.27晚上授课(19:00-22:00)
2026.05.28晚上授课(19:00-22:00)
2026.05.30全天(9点-11-2点-5点)
2026.05.31全天(9点-11-2点-5点)
2026.06.03晚上授课(19:00-22:00)
2026.06.04晚上授课(19:00-22:00)
2026.06.05晚上授课(19:00-22:00)
2026.06.06晚上授课(19:00-22:00)
AIDD人工智能药物发现与设计
2026.05.23全天(9点-11点–2点-5点)
2026.05.24全天(9点-11点–2点-5点)
2026.05.30全天(9点-11点–2点-5点)
2026.05.31全天(9点-11点–2点-5点)
2026.06.06全天(9点-11点–2点-5点)
合成生物学与基因电路设计
2026.05.25晚上授课(19:00-22:00)
2026.05.26晚上授课(19:00-22:00)
2026.05.27晚上授课(19:00-22:00)
2026.05.28晚上授课(19:00-22:00)
2026.05.30全天(9点-11点–2点-5点)
2026.05.31全天(9点-11点–2点-5点)
2026.06.02晚上授课(19:00-22:00)
2026.06.03晚上授课(19:00-22:00)
机器学习代谢组学
2026.05.16全天(9点-11点-2点-5点)
2026.05.17全天(9点-11点-2点-5点)
2026.05.18晚上授课(19:00-22:00)
2026.05.19晚上授课(19:00-22:00)
2026.05.20晚上授课(19:00-22:00)
2026.05.21晚上授课(19:00-22:00)
2026.05.23全天(9点-11点-2点-5点)
深度学习在多组学融合中的应用
2026.05.16全天(9点-11点-2点-5点)
2026.05.17全天(9点-11点-2点-5点)
2026.05.18晚上授课(19:00-22:00)
2026.05.19晚上授课(19:00-22:00)
2026.05.20晚上授课(19:00-22:00)
2026.05.21晚上授课(19:00-22:00)
2026.05.23全天(9点-11点-2点-5点)

报名费用及福利
AI蛋白质设计、
公费价:6380元 自费价:5880元
人工智能构建虚拟细胞、
AI抗菌肽设计、
CADD计算机辅助药物设计、
AIDD人工智能药物发现、
合成生物学与基因电路设计、
机器学习代谢组学、
深度学习在多组学融合中的应用、
每人每班公费价:5880元
每人每班自费价:5480元
优惠福利如下:
优惠一:报二赠一:10880(可选三门课程)
优惠二:报三赠一:13880(可选四门课程)
优惠三:报四赠二:18880(可选六门课程)
优惠四:全报25880(两年内可参加本公司举办的任何课程,不限次数及课程,包括之后新课)
限时福利:报名成功后转发朋友圈或转发50人以上群聊即可获得300元现金红包(只限前15名)
课程福利:报名缴费后可获取往期课程回放(包含全套课程回放和课件资料ppt)提供学员课前预习
报名费用可开具正规报销发票及提供相关缴费证明、邀请函,可提前开具报销发票、文件用于报销 。报名缴费后即可获得全套预习资料供大家课前准备
证书:参加培训并通过考试的学员,可以申请获得工业和信息化部工业文化发展中心颁发的“工业强国建设素质素养提升尚工行动”岗位能力适应评测证书。该证书可在中心官网查询,可作为能力评价,考核和任职的重要依据。评测证书查询网址:www.miit-icdc.org(自愿申请,须另行缴纳考试费500元/人)

SIMPLICITY
官方联系人

本内容为受托转发,出于信息分享目的发布。文中观点、陈述及宣传内容均来自原发布方,并不代表本人立场或意见。如有任何疑问或需进一步核实,请联系原主办单位或讲师团队。本人不参与课程组织、招生、费用收取等事宜,敬请知悉。