 夜雨聆风
夜雨聆风





药物设计师绝不会告诉你的蛋白下载秘密

做药物设计、分子对接,第一步就是下载蛋白结构。但同一个蛋白可能有几百个PDB文件,到底该选哪个?选错了,后续计算结果全是“垃圾”,白白浪费时间。
别急,跟着这三步走,轻松搞定。
01 精准搜索,不迷路
打开PDB官网(www.rcsb.org),在搜索框输入目标蛋白名称,比如“AKT1”。所有已解析的结构会立刻列出来,按时间、物种、表达系统等分类清晰。别被几百条结果吓到,咱们下一步就开始“大浪淘沙”。
02 科学筛选,看三点
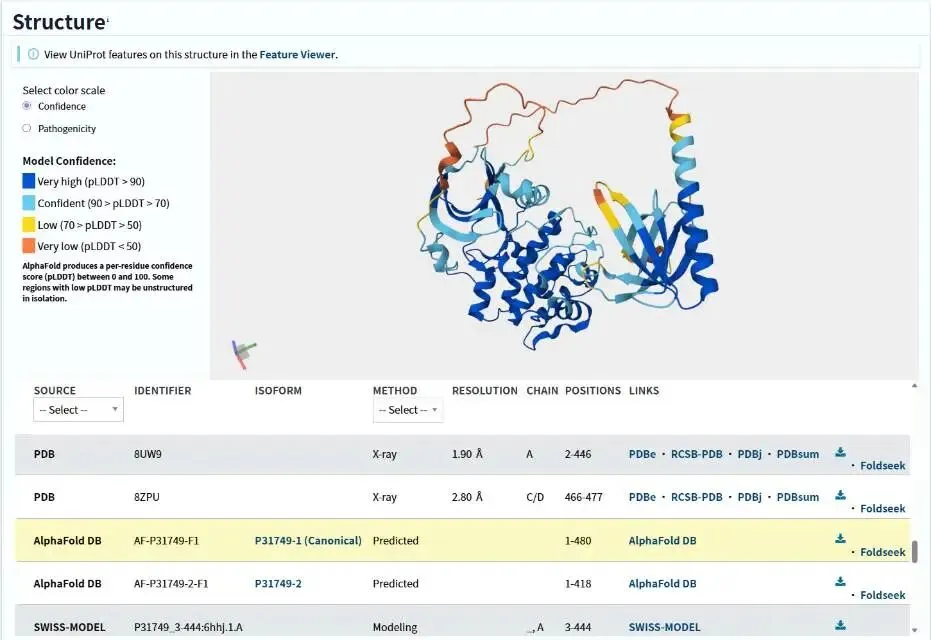
1.AlphaFold预测结构(可选但好用)
如果实验结构不够好,可以先看AlphaFold预测结果。结构模型会用颜色标注可信度:从蓝色(高可信)到红色(低可信)。优先选择蓝色区域覆盖关键活性位点的模型。
2.分辨率——数值越小越清晰
分辨率是实验结构的“清晰度”。记住一个简单规则:≤2.0 Å 非常优秀,2.0–2.5 Å 良好可用,>3.0 Å 要谨慎使用。对于分子对接,尽量选2.5 Å以下的结构。
3.发表年份——越新越好
新结构往往采用更先进的解析方法(如冷冻电镜、更精细的X射线衍射),模型质量更高。优先选近5–10年发表的结构。
03 选对类型——这是最关键的诀窍
很多新手栽在这一步。
如果你是做药物设计、虚拟筛选、分子对接,一定要选择带有配体的“蛋白-配体复合物”结构(PDB文件中会标注存在小分子配体,如抑制剂、底物、辅因子等)。
为什么这么重要?
有配体的结构:配体结合后,蛋白的活性口袋会诱导适配,形成一个闭合、紧致、形状规整的“可成药构象”。这种构象才是药物分子真正结合的“锁定状态”。
没有配体的结构(空结构/Apo结构):活性口袋往往是开放、松散、甚至部分坍塌的。用这种结构做分子对接,你会发现很多“假阳性”——明明不结合的分子也能塞进去,结果完全不可靠。
一句话总结:有配体,才成药;选错型,白辛苦。
小张说模拟
如果你有靶点、有候选分子,却卡在如何进一步提升层级,分子对接+分子动力学就是现在的黄金搭档。小张这里有成熟服务器和对接流程——easy2md平台,可以快速完成结合模式预测与亲和力评估,协助你把文章再往上推一档。