 夜雨聆风
夜雨聆风





一文读懂基因编辑神器之碱基编辑器:知其然、知其所以然
如果想成为基因编辑的专家,那么,一定要将本篇文章同 一口气聊一聊基因编辑(ZFN, TALEN, CRISPR Cas9) 和 一文读懂CRISPR Cas的编辑工具(Cas9, Cas12a, Cas12b, Cas12e, Cas13a, Cas13b, Cas13d) 一并阅读,再结合 3-6个月:如何高效的成为某个领域的专家(人人都会的实操版) 这篇方法,即使没有成为真正意义上的专家也能降维打击一般人!
如果说前2篇文章 一口气聊一聊基因编辑(ZFN, TALEN, CRISPR Cas9) 一文读懂CRISPR Cas的编辑工具(Cas9, Cas12a, Cas12b, Cas12e, Cas13a, Cas13b, Cas13d) 讲解的ZFN、TALEN、CRISPR-Cas系列是确定基因序列后的定位识别与切割到定位灵活后的精准切割。前者需要定位的蛋白与核酸内切酶协同、后者则是利用自身具备的靶序列定位sgRNA和切割酶活性,以此进行基因序列的编辑—比如通过基因编辑去掉HIV基因使个体变得正常。
那么,是不是有更加精准的基因编辑工具作用于单个碱基的编辑呢?比如可以作用于单碱基突变引起的遗传性耳聋、镰状细胞贫血等遗传性疾病的治疗当中。于是碱基编辑器问世了。
一、基础知识
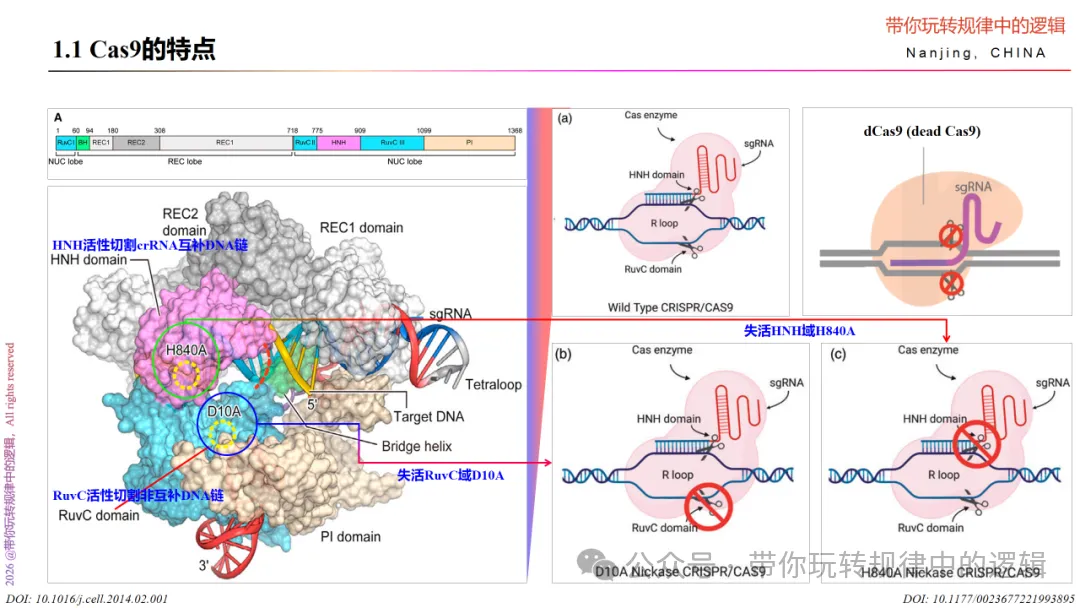
所谓的碱基编辑器,是在Cas9蛋白的基础上,通过人为改造融合其它蛋白而获取的碱基编辑工具。因此,想要搞清楚碱基编辑器,就需要先掌握Cas9的特点,才能更好地理解碱基编辑器的原理。当然,也可以基于其它Cas蛋白的特点,进行工程化改造编辑工具。
首先,从之前的文章,我们了解到,Cas9蛋白有两个酶切结构域,分别是HNH结构域、和RuvC结构域。HNH结构域活性是切割crRNA互补的DNA链的,即靶基因序列所在的DNA链;而RuvC结构域活性则是切割非互补DNA链的。
其次,我们也知道,控制上述两种切割活性的蛋白结构域位点信息。控制HNH结构域活性的是H840A,控制RuvC结构域活性的是D10A。所以,如果通过改造蛋白活性位点的方式,去除H840A,则获得失活HNH结构域活性但有RuvC结构域发挥作用的Cas9,称之为H840A Nickase Cas9;反之,若去除D10A,则获得失活RuvC活性但有HNH活性的Cas9,即D10A Nickase Cas9。但如果把这两个活性全都去除—既没有HNH活性也没有RuvC活性,那么,只能在sgRNA的作用下识别基因序列而无法切割,称之为dead Cas9。
由此,将上面的失活其中一个切割活性的H840A Nickase Cas9或D10A Nickase Cas9,简称为nCas9;两个割切活性都失活的dead Cas9简称为dCas9。
接下来的碱基编辑器,是一直围绕着上述Cas蛋白与特定的具有碱基转换的酶/蛋白的共同作用展开的。
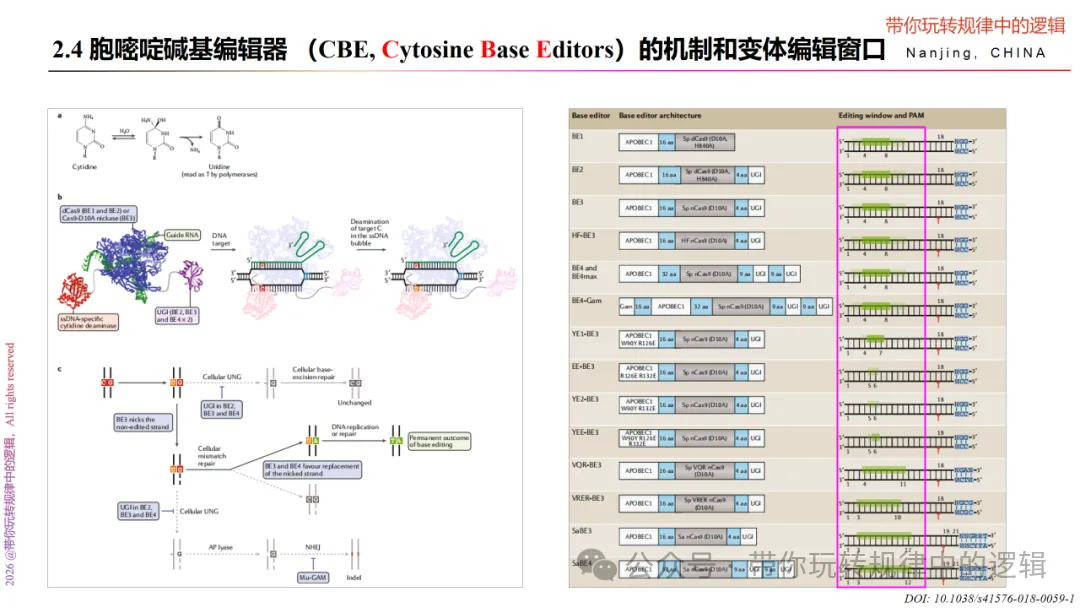
二、胞嘧啶碱基编辑器(CBE, Cytosine Base Editors)
胞嘧啶碱基编辑器 ,可谓是碱基编辑器开发的代表作之一。根据对其的研究和优化改造,目前已经到第四代CBE4(BE4)。
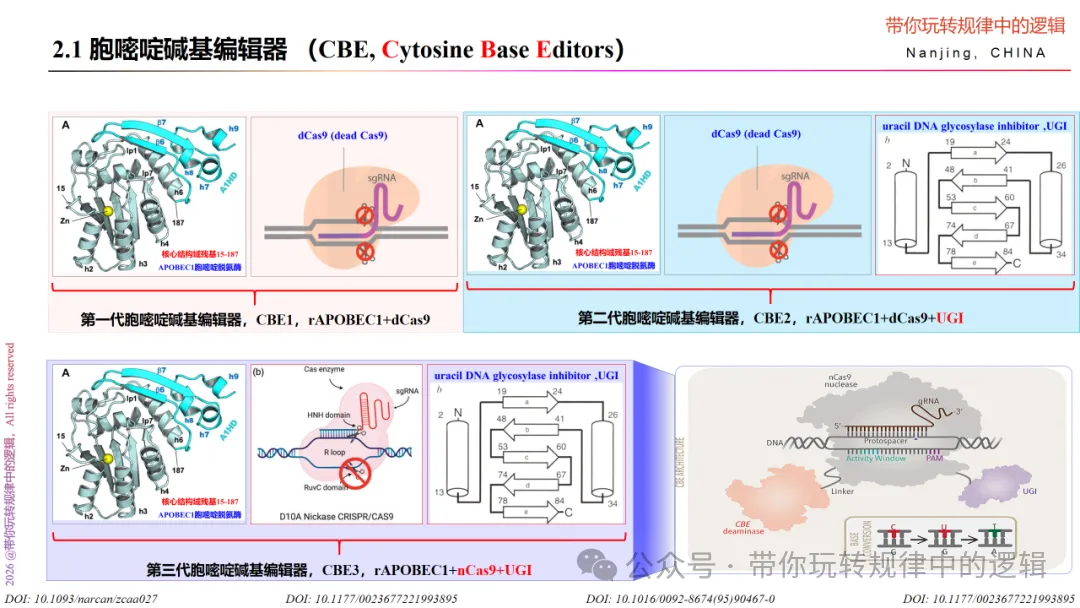
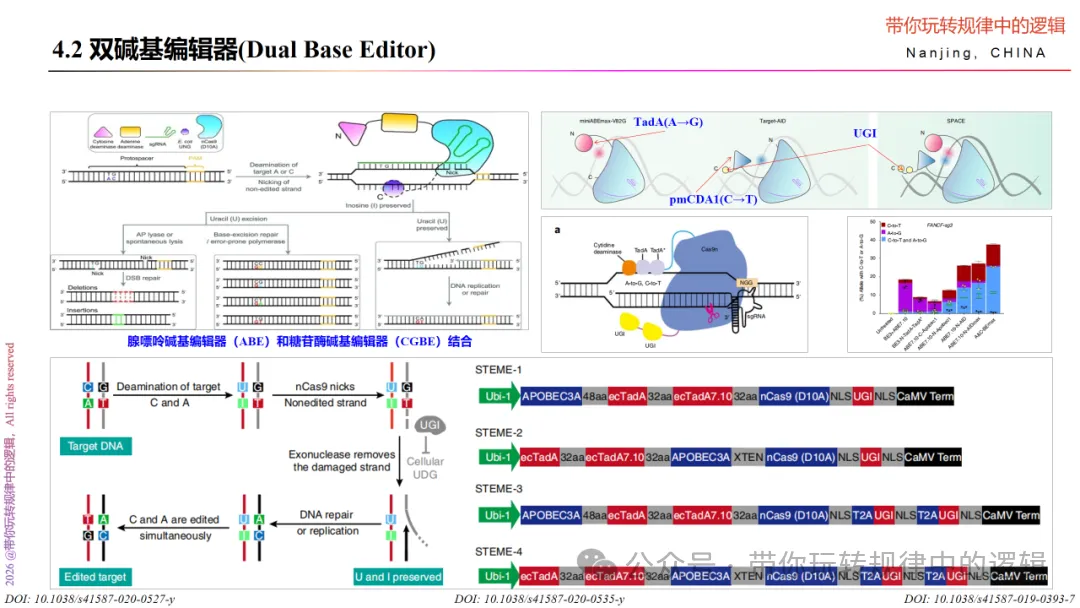
第一代胞嘧啶碱基编辑器 CBE1主要是利用胞嘧啶脱氨酶(APOBEC1)、Linker、dCas9的融合系统。在dCas9靶向特定序列后,胞嘧啶脱氨酶行使将sgRNA及基因组DNA形成的R-loop区的ssDNA处的R-环内的胞嘧啶(C)脱氨成为尿嘧啶(U)的作用,然后在DNA聚合酶的作用下识别尿嘧啶U碱基为胸腺嘧啶T,继而达到碱基由C→T 转换的目的。但是,该种系统的编辑效率不足40%,原因在于被胞嘧啶脱氨酶脱氨转化的中间体尿嘧啶U,会在尿嘧啶DNA糖基化酶(Uracil DNA N-Glycosylase, UNG)的存在下去除,然后在碱基修复途径恢复至C碱基,即 U .G→ C .G。
为了解决尿嘧啶DNA糖基化酶的影响,在第一代CBE1系统中引入了尿嘧啶糖基化酶抑制剂(Uracil DNA Glycosylase Inhibitor, UGI)来消弱尿嘧啶DNA糖基化酶的作用,使得转化的U碱基存在的时间延长、提升编辑的效率,此为第 二代胞嘧啶碱基编辑器 CBE2。
又因为细胞自身具有碱基错配修复机制路径(Mismatch Repair, MMR),而该路径的作用方式是先识别DNA链上的错配碱基,再以含有脱氨酶作用产生的尿嘧啶U的DNA链为模板,对非U碱基DNA链进行修复合成。因此,要保证该机制顺利进行,就需要在非编辑链上产生单链缺口(nick)。从上面介绍Cas9蛋白的信息,我们知道,通过突变D10A失活RuvC活性是解决该问题的办法。于是 第三代胞嘧啶碱基编辑器 诞生了—在nCas9的N端通过Linker融合胞嘧啶脱氨酶、在nCas9的C端引入尿嘧啶糖基化酶抑制剂,再加上DNA存在细胞核中,需要核定位信号NLS,于是形成了APOEBEC1 + Linker + N端–nCas9–C端 + UGI + NLS的系统。
经由第三代胞嘧啶碱基编辑器编辑的产物其实并非是唯一的。主要是尿嘧啶DNA糖基化酶会将转化的中间体U碱基切除形成AP位点,可在DNA复制时将C碱基转化为其它碱基,或在AP裂解酶作用下产生插入缺失,进而形成双链DNA断裂的情况。为降低或避免该种情况出现的频率,在第三代胞嘧啶碱基编辑器基础上继续引入尿嘧啶糖基化酶抑制剂(Uracil DNA Glycosylase Inhibitor, UGI)增强对尿嘧啶DNA糖基化酶的抑制,即含有2×UGI的 第四代胞嘧啶碱基编辑器 。
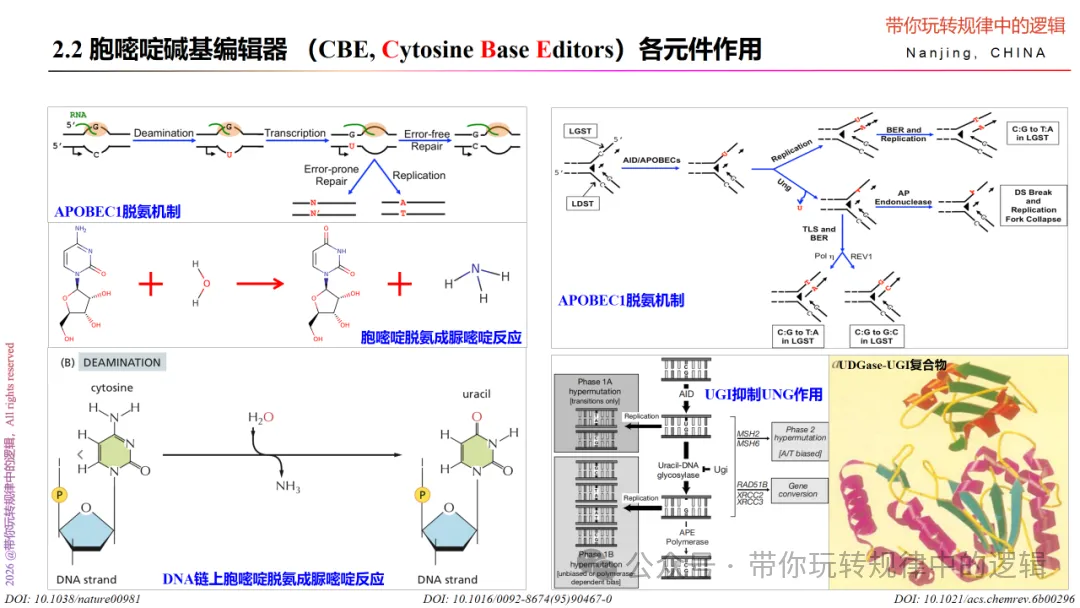
那么,该如何构建胞嘧啶碱基编辑器呢?从上面的讲解中,我们不难看出 胞嘧啶碱基编辑器的组成核心三大部分: 胞嘧啶脱氨酶、nCas9、尿嘧啶糖基化酶抑制剂。其中,胞嘧啶脱氨酶位于nCas9的N端,由Linker连接,确保融合蛋白的灵活性;nCas9保证HNH切割活性,还需要sgRNA的靶向基因的作用,就少不了设计sgRNA了,如何设计sgRNA,参考 链接 一口气聊一聊基因编辑(ZFN, TALEN, CRISPR Cas9) 中CRISPR Cas9部分的sgRNA设计的内容 即可。
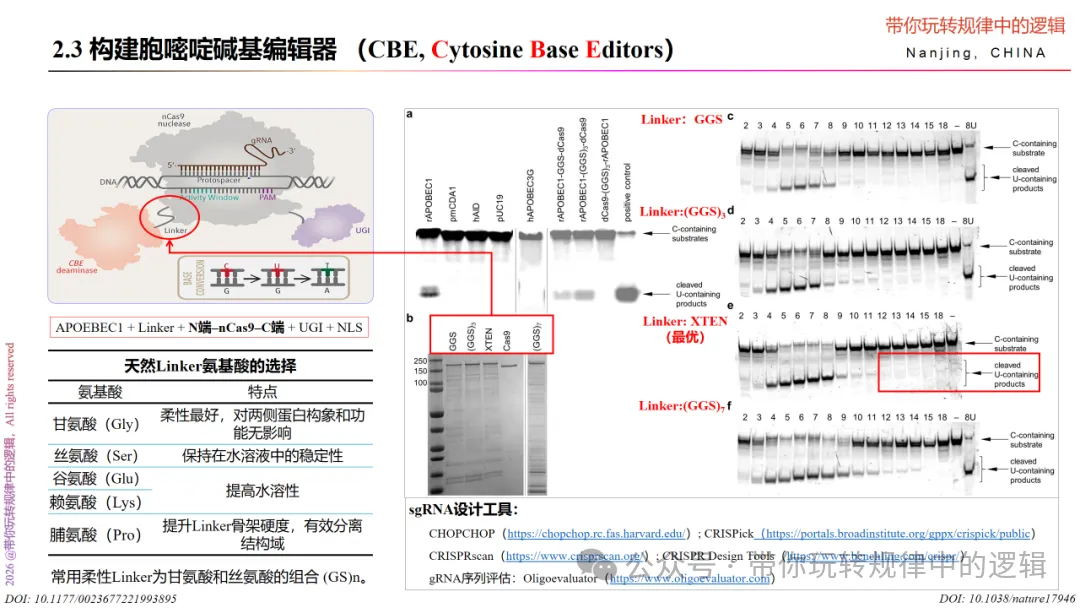
既然知道了胞嘧啶碱基编辑器CBE的核心三部分组成,以及各个核心的作用特点,那么就可以分别就核心三部分进行改进提升编辑性能,比如突变胞嘧啶脱氨酶的位点、更换nCas9蛋白来源或突变位点、改变linker的长度等等获取适合研究目的的胞嘧啶碱基编辑工具。
此外,也可利用dCas9的特点,开发 表观基因组编辑器 —不改变DNA系列,但通过影响蛋白质与DNA的相互作用来修饰基因表达。比如dCas9/gRNA复合物被导向基因启动子或转录增强子中的某个序列,通过空间位阻干扰通常与该序列相互作用的因子,从而影响基因表达(CRISPR 干扰);或将dCas9与能主动抑制基因表达的结构域融合,通过改变局部染色质结构进而影响DNA序列对转录机制的可及性,可实现更强的基因敲除效果;或将dCas9与甲基转移酶或去甲基化酶结构域融合,可实现更持久的表观基因组编辑。

三、腺嘌呤碱基编辑器 (ABE, Adenine Base Editors)
详细了解了胞嘧啶碱基编辑器,相信对腺嘌呤碱基编辑器的掌握会更加高效。
腺嘌呤碱基编辑器相较于胞嘧啶碱基编辑器,其系统主要是ecTadA*+ Linker(XTEN) + nCas9(D10A)。采用的是腺嘌呤脱氨酶对腺嘌呤碱基A脱氨变为肌苷I,而后肌苷I在DNA复制时被当作鸟嘌呤G,实现A→G的转换。
但是一般的腺嘌呤脱氨酶是无法直接作用于单链DNA模板上A碱基的。因此,需要对腺嘌呤脱氨酶进行工程化改造。如对大肠杆菌腺嘌呤脱氨酶TadA改造成TadA*以作用于ssDNA的A碱基。
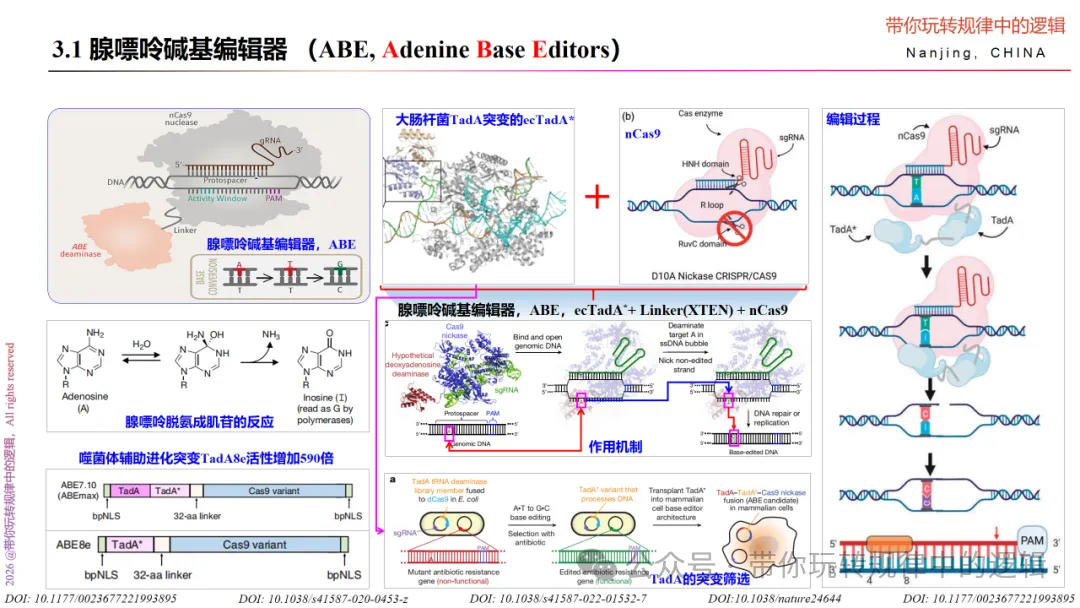
根据胞嘧啶碱基编辑器要解决尿嘧啶DNA糖基化酶的影响,也需要对腺嘌呤碱基编辑器的甲基嘌呤DNA糖基化酶的影响做验证。通过在nCas9引入抑制N-甲基嘌呤 DNA 糖基化酶(N-methylpurine DNA glycosylase, MPG or AAG),编辑产物的纯度并没有发生改变,因此,在构建腺嘌呤碱基编辑器时可省去MPG的部分。
因此,想要提升腺嘌呤编辑器的编辑效率,则需要在腺嘌呤脱氨酶上下功夫。比如调整腺嘌呤脱氨酶二聚体的位置、密码子优化、改变核定位信号位置等,以提升编辑效率或扩大编辑窗口。如通过突变体库筛选出高活性的TadA8e。
该如何构建腺嘌呤碱基编辑器呢?方法和上述的胞嘧啶碱基编辑器如出一辙,不再赘述。
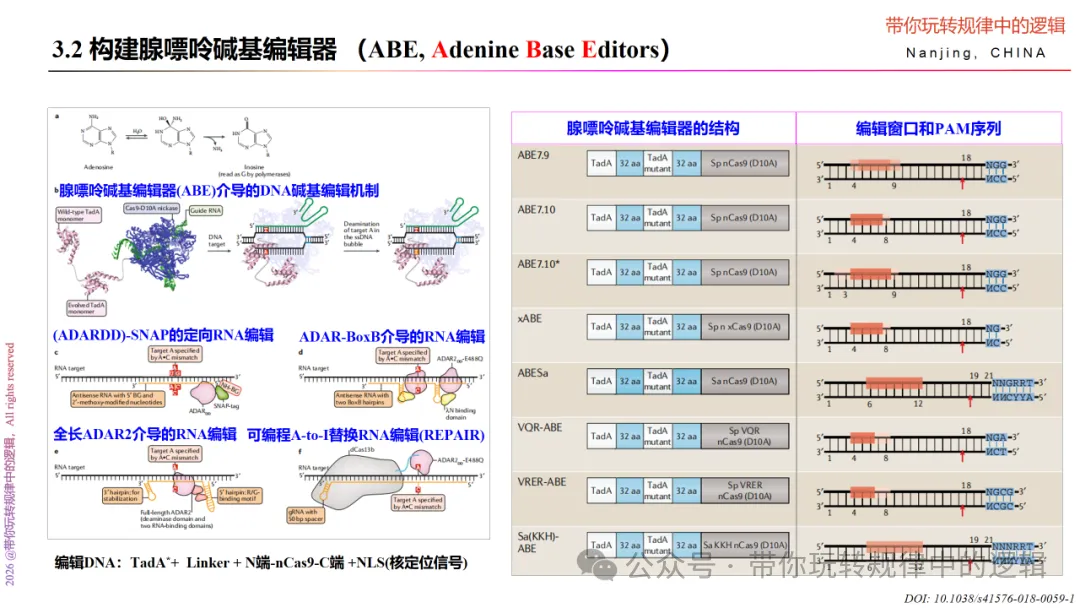
此外,也可通过替换nCas9为Cas13b,再与RNA腺苷脱氨酶(Adenosine deaminase acting on RNA,ADAR)融合,通过Cas13b-crRNA进行定位到靶向位点,并利用ADAR催化A-I的转化,进而实现对RNA的编辑。
四、碱基编辑器的衍生(gTBE、gCBE、GBE、AYBE、CGBE、双/三碱基编辑器)
对胞嘧啶碱基编辑器、腺嘌呤碱基编辑器了解清楚后,在二者的基础上衍生的其它碱基编辑器就更加容易了—其实就是更改编辑的酶达到特定的碱基变换目的。
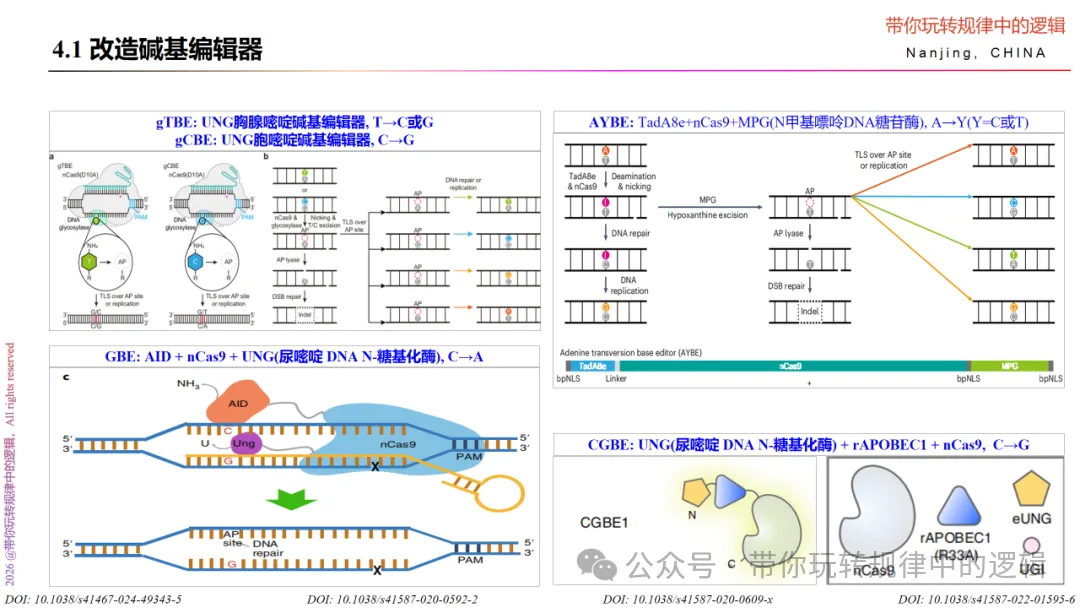
gTBE: 工程化尿嘧啶DNA糖基化酶(UNG)胸腺嘧啶碱基编辑器,nCas9+UNG。在sgRNA特异性结合靶DNA上形成R-loop结构,暴露单链DNA;工程化改在的尿嘧啶DNA糖基化酶恰好识别单链DNA上的胸腺嘧啶T碱基,进而切除形成AP位点;而细胞内的碱基切除修复机制的作用下,转换为C或G碱基。
gCTB: 工程化尿嘧啶DNA糖基化酶(UNG)胞嘧啶碱基编辑器。同gTBE,gCBE作用于C碱基上,通过工程化的糖基化酶去除胞嘧啶,在修复时DNA聚合酶的作用下掺入鸟嘌呤G,实现C→G的转换。
GBE: AID + nCas9 + UNG(尿嘧啶 DNA N-糖基化酶)。AID胞苷脱氨酶将胞嘧啶C脱氨为U,而后UNG识别并切除U碱基,形成AP位点;细胞内修复时,DNA聚合酶根据互补链模板碱基,修复时引入腺嘌呤A配对,成功将C碱基转化为A碱基。
CGBE: UNG(尿嘧啶 DNA N-糖基化酶) + rAPOBEC1 + nCas9。rAPOBEC1将C碱基脱氨转化为U碱基;UNG识别U碱基并去除形成AP位点;利用DNA修复时引入G碱基插入原本C碱基的位置,进而将C替换G。
AYBE: TadA8e+nCas9+MPG(N甲基嘌呤DNA糖苷酶)。利用高活性嘌呤脱氨酶将A碱基脱氨为肌苷I,N-甲基嘌呤DNA糖苷酶(MPG)识别并切除肌苷I形成AP位点;以编辑链的I为模板,插入C(I-C配对),最终实现 A→C,或随机插入T(保留原配对),导致 A→T的转换。
了解了各种单碱基编辑器,双碱基编辑器和和三碱基编辑器就是在碱基转换的酶做了二元组合与调整。比如 双碱基编辑器 是腺嘌呤碱基编辑器(ABE)和糖苷酶碱基编辑器(CGBE)结合,既具备TadA腺嘌呤脱氨酶将A转化为G的能力,又具备胞嘧啶脱氨酶将C转化为G的能力。
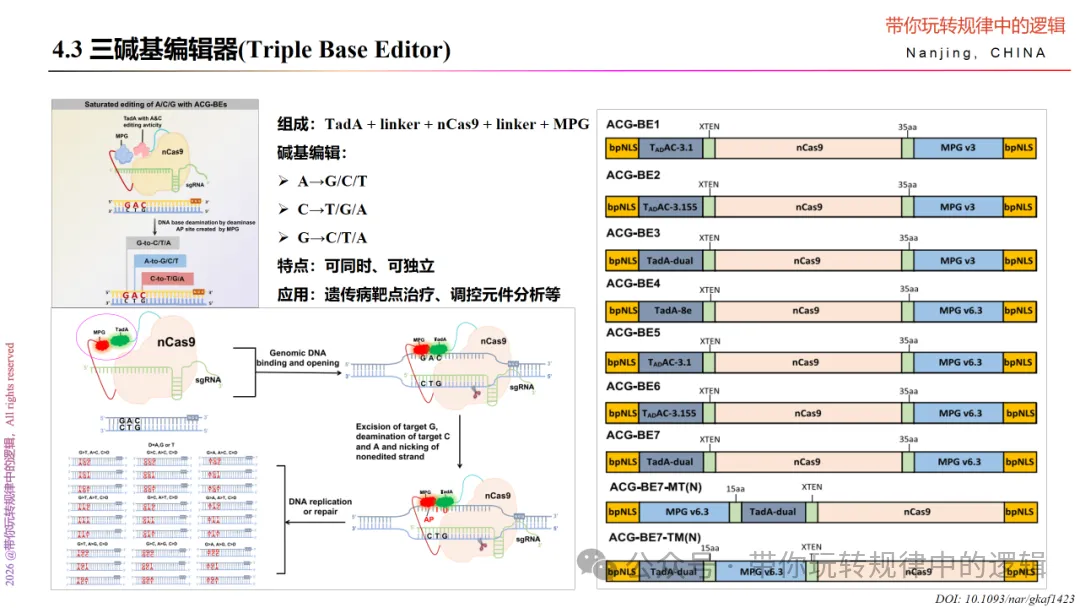
三碱基编辑器 ,则是融合了高 A/C 催化活性的TadA腺嘌呤碱基编辑器与工程化的N-甲基嘌呤DNA糖基化酶(N-methylpurine DNA glycosylase, MPG),催化腺嘌呤(A)、胞嘧啶(C)和鸟嘌呤(G)三种碱基的饱和突变,可实现单独或同时A→G/C/T、C→T/G/A、G→C/T/A等多种碱基转换或颠换,覆盖64个遗传密码子中的63个,能产生所有19种氨基酸突变及终止密码子。该种类型的碱基编辑器在蛋白定向进化、遗传筛选等疾病靶点治疗方面有着重要应用价值。
五、引导编辑器
上述的单碱基编辑器、双碱基编辑器、以及三碱基编辑器,都会在后续的细胞内修复或者DNA复制机制进行碱基的替换或转换时产生副产物、以及脱靶的比率。那么,可否开发出一种工具来解决此类问题呢?于是引导编辑器被设计出来了。
引导编辑器 是由 三部分组成 的,分别是nCas9(H840A)、去除RNaseH活性的MMLV逆转录酶、以及pegRNA(Prime Editing Guide RNA, pegRNA)。其中,引导编辑器是在nCas9的C端融合了MMLV逆转录酶构建的编辑系统;而pegRNA则是包含与靶基因互补的Spacer序列、nCas9的RNA支架结构Scaffold、编辑位置的序列、以及逆转录时引物结合位点序列PBS(primer binding site, PBS)。
引导编辑器编辑的整个过程 分为三步:首先,在pegRNA的引导下nCas9精准定位到目标位置所在的互补链上,并在目标位置所在链上切割形成切口;其次,被切开的目标DNA单链从双螺旋中释放出来,与pegRNA的PBS序列配对结合,此时,逆转录酶MMLV以切割的单链DNA链作为引物,合成一段带有正确序列的新的DNA序列;最后,新和成且携带编辑信息的DNA单链—即编辑后的新的DNA链嵌入原来的DNA双螺旋中,通过模板置换取代原始DNA链。而后在细胞自身的DNA修复机制下,另一条链被修正成和新合成的链完全互补的序列,实现被编辑序列的保留。
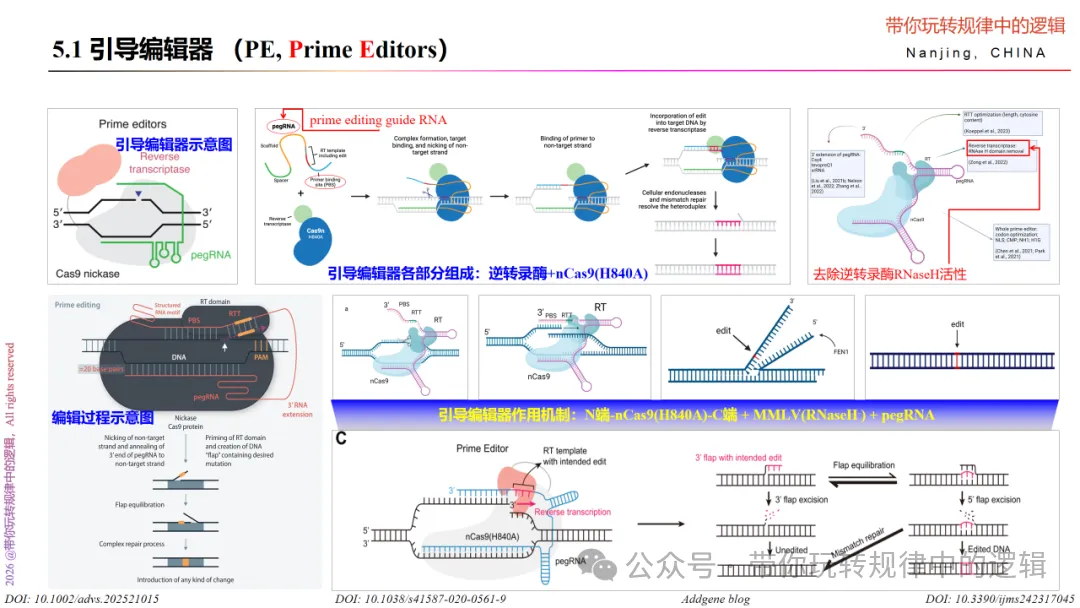
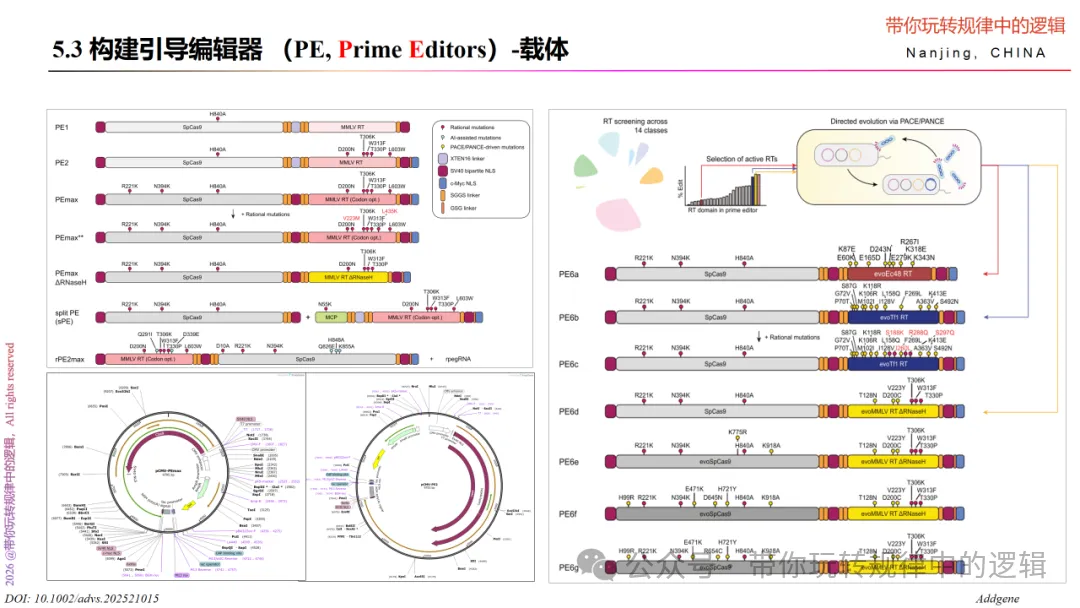
由此,将上面的引导编辑器定义为PE1版本,那么,就可以分别针对其中的nCas9、MMLV(RNaseH – )、以及pegRNA序列等三者分别做改进来提升整个引导编辑器的编辑效率。比如提高逆转录效率的PE2系统;额外引入sgRNA将nCas9引导至编辑位置附近使编辑后的链作为模板驱动错配修复的PE3系统;又如两个PE对相反链进行互补编辑的Twin PE系统等。

既然,引导编辑器可以通过改进组成的各部分的信息达到编辑的目的。该如何 构建引导编辑器 呢?主要分为两部分,一部分是 设计pegRNA序列 ,一部分是构 建PE系统的载体 。
首先,pegRNA序列的组成 ,我们在本部分开始已经说过了。那么就需要在确定目标基因的序列后,分别设计PBS和逆转录编辑序列的信息了。具体的可以参考下面这张图的提供的设计思路以及原文。当然,也可以通过PEGG网址指示来设计出适合自己实验目的的pegRNA序列,网址链接如下: https://pegg.readthedocs.io/en/latest/ 。另外,《Protocol for the design, conduct, and evaluation of prime editing in human pluripotent stem cells》(DOI: 10.1016/j.xpro.2023.102583)也是值得借鉴的。

其次,是载体部分。 载体可根据已经报道的PE系统的设计来进行个性化改造,也可以购买商业化的载体用于实验。载体构建可参考文献《Prime editing: Mechanism insight and recent applications in plants》(DOI: 10.1111/pbi.14188),或者商业化载体网址,如 https://www.addgene.org/crispr/prime-edit/ ,涵盖哺乳动物、细菌、果蝇、植物、空载等载体序列可供选择。
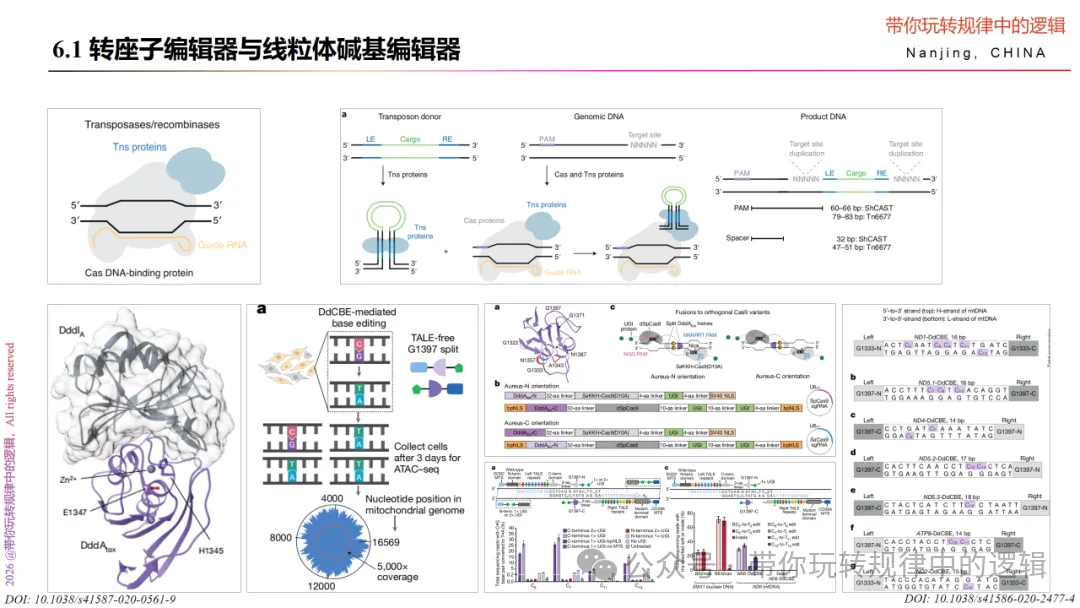
六、转座子编辑器和线粒体DNA编辑器
相较于常见的碱基编辑器,转座子编辑器和线粒体DNA编辑器也有异曲同工之处。
Cas转座子编辑器 由Cas蛋白和转座酶两部分主要组成。当然也离不开编辑序列,由LE+“转运”DNA+RE序列组成。整个编辑序列与转座酶蛋白Tns结合。Cas蛋白依赖PAM序列、sgRNA序列被引导至目标位点。Cas结合使转座酶蛋白定位于目标序列,并促进编辑序列DNA在靶位点的整合。靶位点被复制并位于整合的LE-转运DNA-RE序列两侧。形成新的编辑产物。
DDCBE: 线粒体DNA编辑器,由双链DNA胞苷脱氨酶DDDA和转录激活因子TALE与UGI组成。TALE序列作为定位元件,特异性识别并结合线粒体DNA目标序列,引导编辑器至特定位点。DDDA可直接作用于双链DNA(dsDNA),无需解旋双螺旋结构,催化胞嘧啶(C)转化为尿嘧啶(U)。UGI抑制细胞内尿嘧啶-DNA糖基化酶的活性,防止编辑后的尿嘧啶(U)被识别并修复回胞嘧啶(C)。在这三者的额作用下达到C-G碱基对向T-A碱基对的转换的目的。此外,DDCBE系统还需要线粒体基质导入序列,确保编辑器能穿过线粒体双层膜进入线粒体基质中,从而作用于线粒体DNA上。
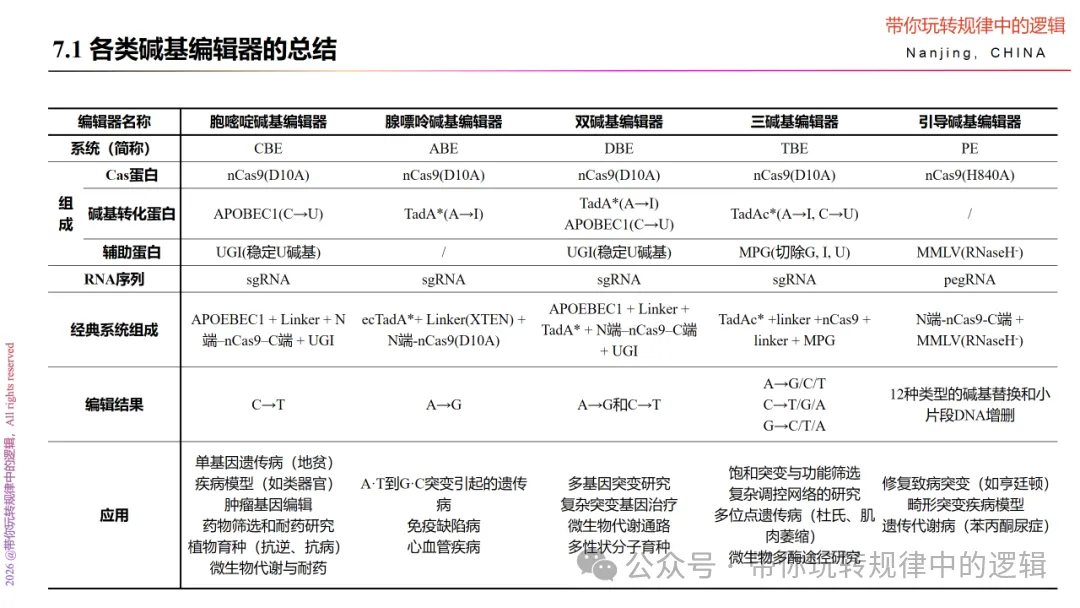
七、总结各类碱基编辑器和思考
根据以上对各类碱基编辑器的讲解,不同的碱基编辑器都是依赖特定的碱基转换酶或者组合来达到碱基编辑的目的的。当然,引导编辑器是不涉及碱基转换酶的,取而代之的是通过逆转录酶引入待替换的碱基序列。
基因编辑从2010年爆发后,研究和应用可谓如火如荼。科学家或者研究工作者无一不在探寻高靶向率且行之有效的基因编辑工具。有3个问题是亟待解决的:(1)临床商业化,无论是伦理、法律法规、临床资源和标准等都是需要国内外多行业人士共通共识的;(2)工具数据库,编辑酶、序列、疾病模型、RNA序列设计等多板块的有效数据管控和使用,以及在大模型下的数字孪生预测,是未来发展需要考虑的问题 生物行业未来5-10年的发展蓝图:从未来看现在 ;(3)聚焦方向和实用性。如PE引导编辑器在植物方面的编辑效率低,是否能调整至其它编辑器或者应用等。这样才能有一个基因编辑工具跑通从研发-临床-商业-法规-医保的等全链条的路线,才能积累更多的成功经验用于其它基因编辑工具上。
声明:个人理解,仅用于阅读。欢迎专业人士留言、斧正!