 夜雨聆风
夜雨聆风





整合 AI、物理模型与实验的 IMPECCABLE 工作流:纳摩尔级抑制剂发现

一、研究背景与立题依据
AI 方法的核心短板
生成式 AI 虽能实现大规模分子生成,但其能力受限于训练数据集,仅能在编码空间内进行插值与外推,缺乏对未知化学空间的可靠泛化能力;同时 AI 预测无法实现系统的不确定性量化,输出结果常过度自信,假阳性率高,难以获得药物化学家的信任。
物理基方法的核心短板
基于分子动力学的结合自由能计算、分子对接等方法,预测精度高、可实现严格的不确定性量化,结果可靠性强,但计算成本极高、速度慢,无法实现十亿级化学空间的全量筛选。
二、IMPECCABLE 工作流的整体架构与核心模块
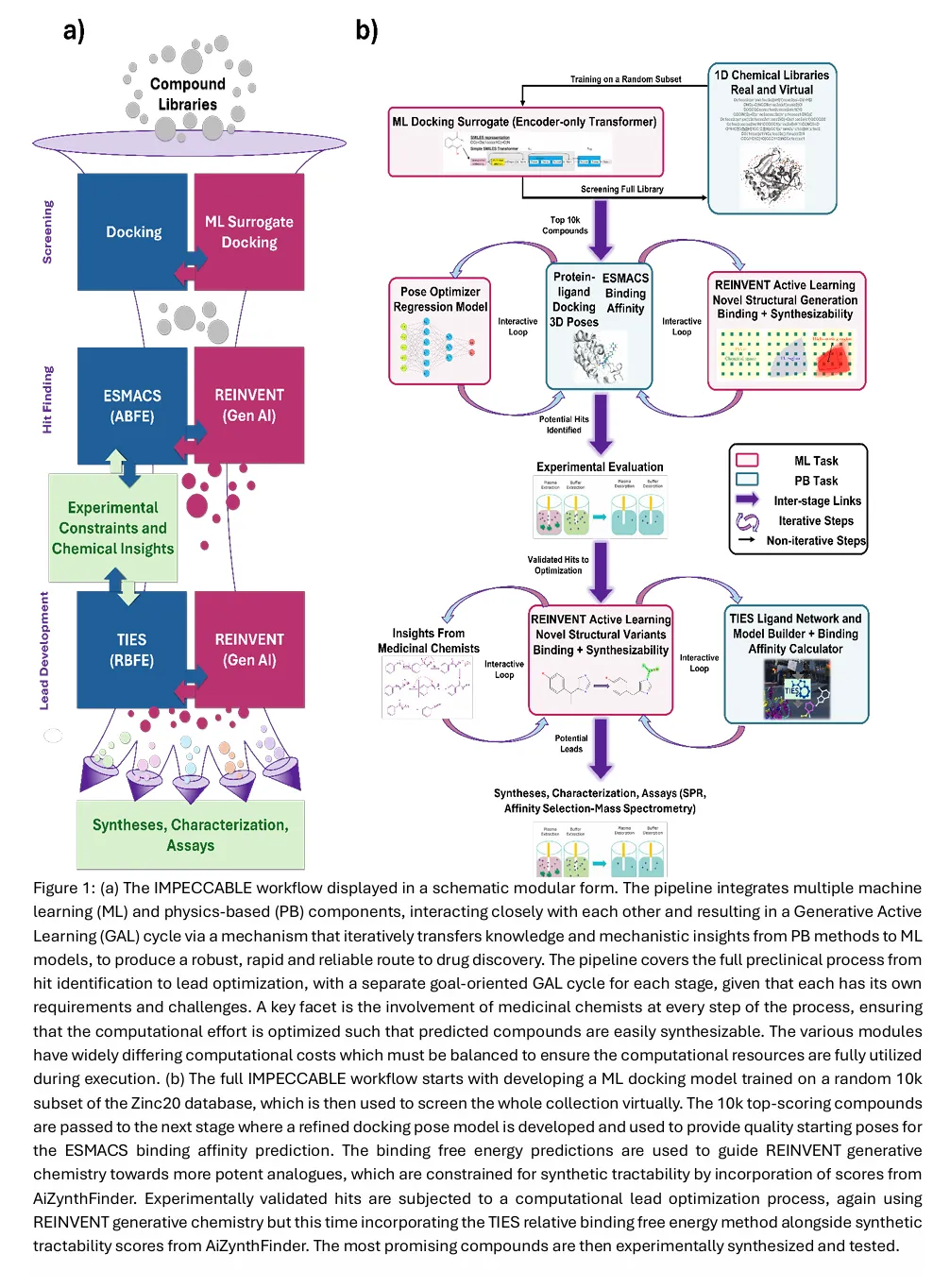
ML 对接替代模型训练:基于 Zinc20 数据库的随机 10k 子集,训练仅编码器 Transformer 模型,作为分子对接的替代模型,实现超高速虚拟筛选。
全库虚拟筛选:用训练完成的替代模型对整个化合物库进行筛选,取评分最高的 10k 个化合物进入下一阶段。
对接构象优化:通过 AI 构象优化器,为筛选得到的化合物生成高质量的蛋白结合构象,作为后续自由能计算的初始结构。
hit 发现 GAL 循环:以 ESMACS 方法计算的绝对结合自由能为反馈信号,引导 REINVENT 生成式 AI 优化分子结构,同时通过 AiZynthFinder 评分约束分子的可合成性,兼顾结构多样性与新颖性。
实验初步验证:对筛选得到的 hit 分子进行合成、结构表征与亲和力检测,检测方法包括表面等离子体共振(SPR)、亲和选择 – 质谱等。
先导优化 GAL 循环:以实验验证的 hit 分子为起点,用 TIES 方法计算的相对结合自由能替代 ESMACS,结合 REINVENT 与 AiZynthFinder,进一步优化分子的结合亲和力、可合成性与成药性。
最终验证:对最优候选分子进行合成、手性纯化与全面的活性评价,完成计算 – 实验的完整闭环。
核心功能模块详解
1. 物理基计算模块
分子对接
采用 OpenEye 科学软件套件完成全流程处理,通过 FixpKa 在 pH 7.4 条件下为化合物分配合适的质子化状态,OMEGA 为每个化合物生成最多 200 个构象,FRED 完成分子对接,以 Chemgauss4 评分对结合构象进行排序。
ESMACS
增强采样的分子力学 / 泊松玻尔兹曼表面积方法,用于 hit 发现阶段的绝对结合自由能(ABFE)计算。通过 Binding Affinity Calculator(BAC)接口对接 AmberTools23 套件,配体采用 GAFF2 力场与 AM1-BCC 原子电荷,蛋白采用 Amber ff14SB 力场,水分子采用 TIP3P 模型;每个蛋白 – 配体复合物设置 10 个重复体,经能量最小化、升温后,执行 4 ns 的生产相分子动力学(MD)模拟,在保证预测精度的同时,适配高通量筛选的算力需求。
TIES
增强采样的热力学积分方法,用于先导优化阶段的相对结合自由能(RBFE)计算。基于炼金术自由能微扰原理,通过集合模拟实现系统的不确定性量化,最大限度降低假阳性 / 假阴性结果,为 GAL 循环提供精准的优化方向。计算前先基于最大公共子结构构建配体扰动网络,采用 AM1-BCC-ELF10 电荷方案分配原子电荷,通过 FEgrow 流程制备蛋白结合态坐标;每个配体设置 5 个重复体,覆盖 13 个 λ 窗口,每个窗口执行 4 ns 生产相模拟,单配体总模拟时长达 260 ns。
2. AI 功能模块
生成式主动学习(GAL)
将基于强化学习的 REINVENT 分子生成模型,与物理基结合自由能计算驱动的迭代主动学习深度融合。hit 发现阶段采用循环神经网络(RNN)先验,实现分子的从头生成;先导优化阶段采用 Libinvent 先验,支持用户固定分子骨架,仅对指定出口向量的 R 基团进行优化。每个循环生成 10000 个(hit 发现)/1000 个(先导优化)候选分子,经分子指纹聚类去除相似结构后,通过复合评分函数筛选;采用贪心采集策略选择 100 个分子执行物理基自由能计算,计算结果用于更新替代模型,形成闭环迭代优化。
对接替代模型
开发了 BERT 类 Transformer 回归模型 Simple SMILES Transformer(SST),直接以 SMILES 字符串为输入,无需繁琐的分子描述符预处理,大幅提升筛选通量。单张 NVIDIA A100 显卡每秒可处理 3100 个化合物,在 Aurora 超算的 256 个节点上,可在 40 分钟内完成 ZINC22 数据库 220 亿个化合物的全量筛选。
构象优化器
自研 AI 模型,以配体 – 蛋白结合构象为输入,基于蛋白 – 配体原子间的相互作用,预测构象的 MMPBSA 自由能评分并排序,筛选出最优结合构象,为后续自由能模拟提供高质量初始结构。
三、工作流的应用与实验验证结果
1. 苗头化合物(hit)发现阶段
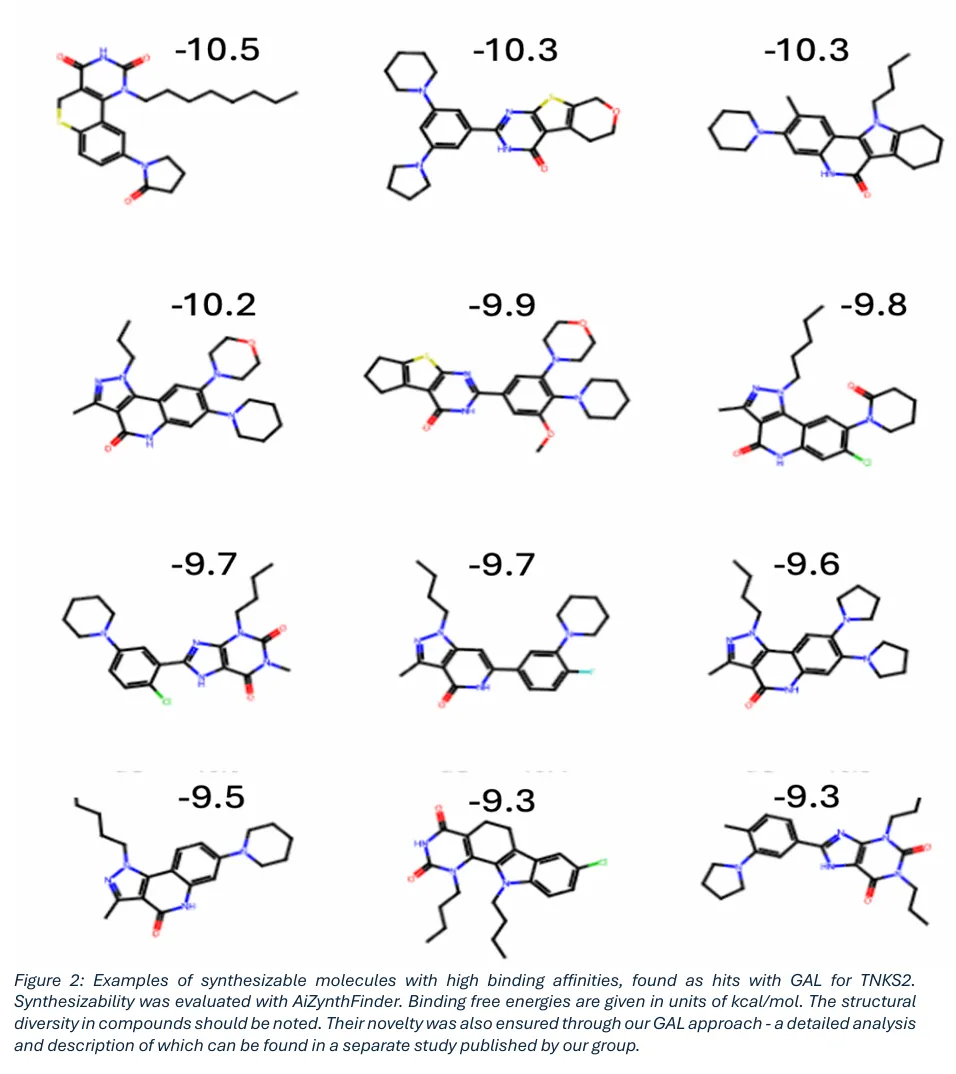
TNKS2 靶点:端锚聚合酶 2(TNKS2)是已验证的致癌靶点,参与端粒维持、有丝分裂、葡萄糖代谢等多种细胞进程。研究以文献报道的 27 个已知 TNKS2 结合剂为种子结构,初始化 GAL 循环,仅 3 次迭代即实现 ESMACS 预测结合亲和力的收敛,得到了一批化学多样性高、可合成性优异的 hit 分子。Figure 2 TNKS2 靶点 GAL 循环发现的可合成高亲和力 hit 分子示例,标注了 AiZynthFinder 评估的可合成性与结合自由能(单位:kcal/mol),直观展示了生成分子的骨架多样性与结构新颖性。
WDR91 靶点:WDR91 蛋白被认为在人体病毒感染宿主应答中发挥关键作用,但其具体生物学功能尚未完全阐明,小分子结合剂是解析其功能的核心工具。该靶点首个小分子结合剂来自 DNA 编码化合物库(DEL)筛选偶联机器学习,先导化合物 1 的 KD 为 6 μM,已有晶体结构明确了结合模式。研究团队先购买了 20 个化合物 1 的结构变体,又在实验室合成了 12 个补全构效关系的分子,SPR 检测结果显示所有化合物的 KD 均≥6 μM,未实现亲和力提升;此前采用的自由能微扰(FEP)方法,预测了一批含 N – 取代硫杂环丁烷二氧化物结构的化合物,但经大量合成努力后,未发现结合力更优的分子,凸显了无系统不确定性量化的模拟方法泛化性差、预测结果不可靠的问题。基于此,研究选择化合物 1 作为先导,进入 IMPECCABLE 先导优化循环。
2. 先导化合物优化阶段
TNKS2 靶点:从 hit 发现阶段的结果中,选取 2 – 苯基 – 6 – 氟喹唑啉 – 4 – 酮为核心骨架,通过 REINVENT 的 Libinvent 先验,分别对 7 位(R1)、苯环对位(R2)两个出口向量进行定向优化。R1 位点完成 5 次 GAL 迭代,R2 位点完成 3 次迭代,结合自由能分布持续向高亲和力方向稳定偏移。
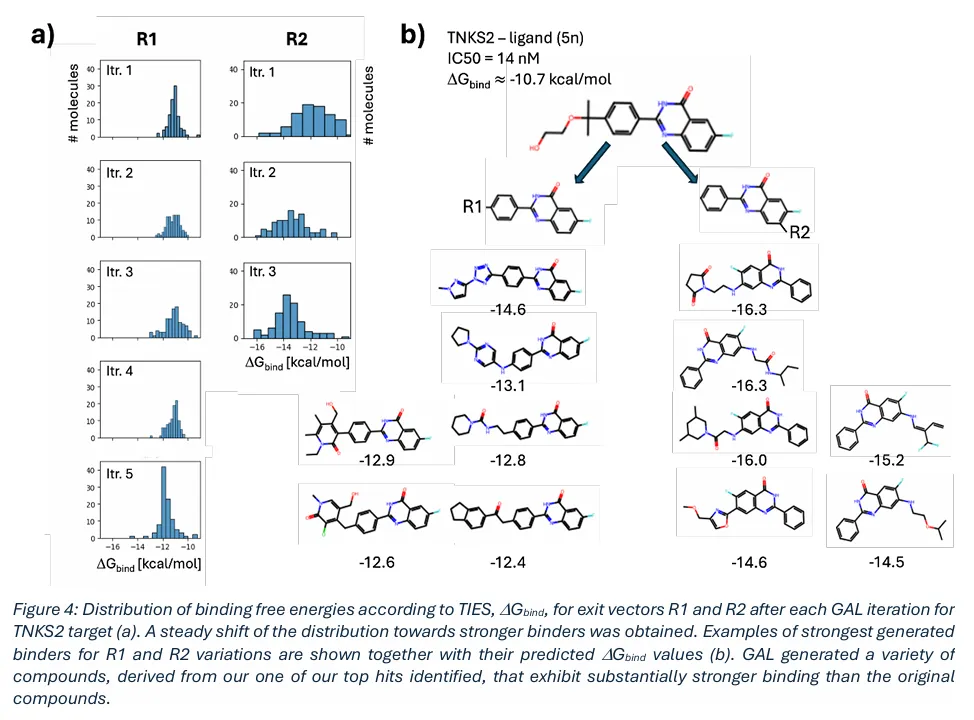
Figure 4 (a) TNKS2 靶点每个 GAL 迭代后,R1、R2 出口向量的 TIES 结合自由能 ΔGbind 分布,清晰显示分布持续向更强结合剂方向偏移;(b) R1、R2 优化得到的最强结合剂示例,以及对应的预测 ΔGbind 值,生成化合物的结合亲和力显著优于原始母体化合物。
WDR91 靶点:以化合物 1 为基础,定义了核心骨架 9 与三个结构出口向量,优先优化 R1(化合物 1 中的氧杂环丁烷)与 R2(化合物 1 中的 3 – 羟基吡咯烷)位点,R3 位点位于狭窄的结合口袋,不适合进行结构修饰。REINVENT 共生成 94 个候选结构,药物化学家基于结构多样性与可合成性,人工筛选出 10-15 个分子进行合成与活性检测。其中化合物 10 在 SPR 实验中(5 次重复)测得 KD 为 0.345 μM,经差示扫描荧光法热位移实验交叉验证,相较于起始化合物 1 实现了 20 倍的亲和力提升,是目前已报道的 WDR91 蛋白最高亲和力结合剂;最终经手性纯化后,化合物 10 的 KD 值为 352 ± 32 nM,达到纳摩尔级强效抑制水平。
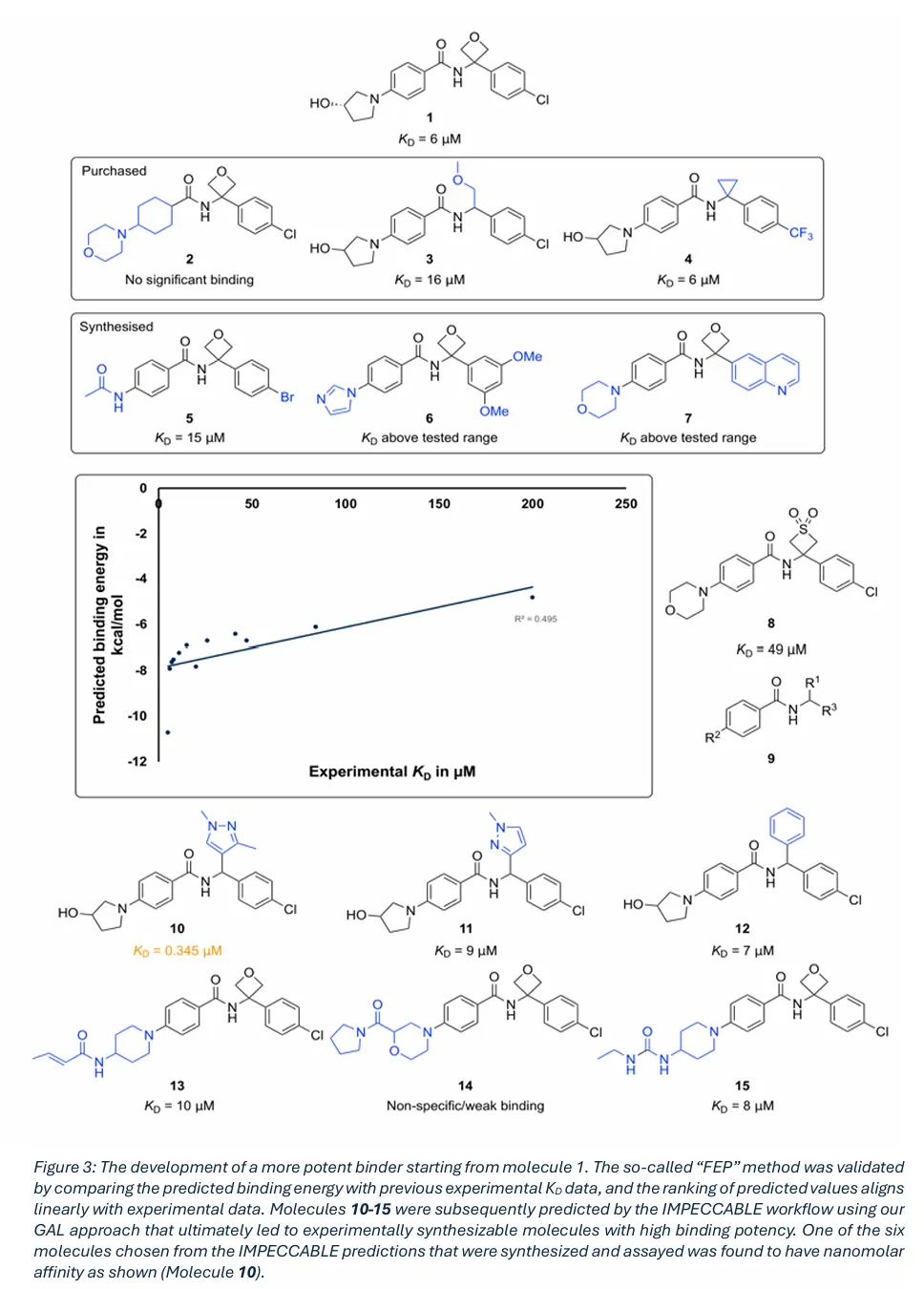
Figure 3 从化合物 1 出发的高亲和力结合剂开发全流程,包含 FEP 方法的验证结果(预测结合能与实验 KD 线性相关,R=0.495)、IMPECCABLE 工作流 GAL 循环预测的分子 10-15,以及合成后实验验证的纳摩尔级亲和力化合物 10 的结构与活性数据。
3. 配体 – 蛋白相互作用的机理分析
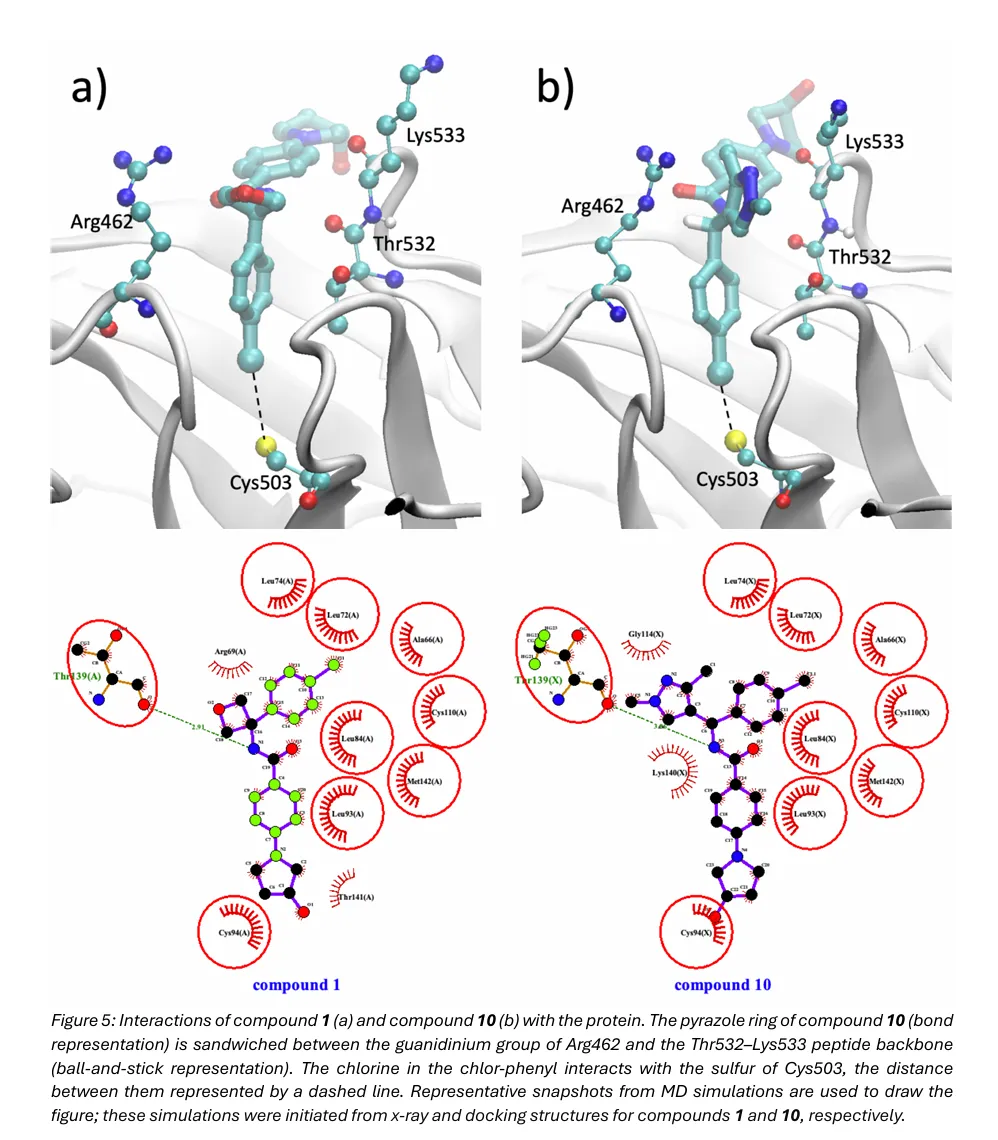
化合物 10 的苯甲酰胺核心酰胺氮,与 Thr532 的主链羰基形成稳定的氢键相互作用。固定不变的 R3 氯苯基上的氯原子,与 Cys503 的硫原子形成相互作用,二者距离为 3.66±0.25 Å,夹角为 133±10°。
亲和力提升的核心来源:R1 位点的吡唑环被精准夹在 Arg462 的胍基与 Thr532-Lys533 的肽主链之间,形成稳定的阳离子 -π- 酰胺三明治堆叠结构,同时利用吡唑环的 π 体系,与两侧分别形成阳离子 -π 相互作用和酰胺 -π 堆叠作用,最大化了与蛋白的结合能。对比化合物 1 的氧杂环丁烷结构,其在结合口袋中发生 90 度旋转,与蛋白残基呈垂直取向,破坏了平面堆叠结构,仅能形成不利的边对面相互作用,因此结合力显著弱于化合物 10。
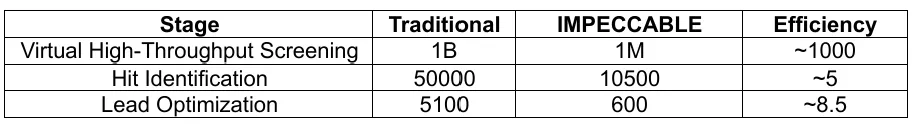
四、工作流的计算效率与性能优势
|
|
|
|
|
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
超算适配与极速处理
全流程在 Frontier 超算上,完成 ZINC20 数据库(约 10 亿化合物)的全流程处理,总墙钟时间仅 25 小时;最新版本处理 ZINC22 数据库(约 370 亿化合物),墙钟时间仅出现边际增加,可在数天内完成从虚拟筛选到候选分子输出的全流程。
高并发与可扩展性
可在 Frontier、Aurora 两台超算上并发执行多个 IMPECCABLE 工作流,相较于配备 1k GPU 的中等规模集群,速度提升至少两个数量级;总吞吐量随并发执行数、化合物库大小呈线性扩展,可同时并行处理多个不同的蛋白靶点。
超高实验成功率
在 WDR91 靶点上,仅合成并测试 6 个预测分子,即发现 1 个纳摩尔级强效抑制剂,远高于传统药物发现的实验成功率。