 夜雨聆风
夜雨聆风





字节AI制药团队出手:AI亲和力预测离真实决策还有多远?


字节AI制药团队出手:AI亲和力预测离真实决策还有多远?

你可能听过这样的说法:AI已经能预测药物分子和蛋白质结合得有多紧了,精度甚至追上了传统物理计算方法。这话不算错,但只对了一半。
4月28日,字节跳动AI制药团队(ByteDance AI Drug Discovery / Anew Therapeutics)在ChemRxiv上发了一篇预印本,干了一件很有意思的事:他们同时站在AI和物理两个阵营,用公开数据和内部真实项目数据,正面回答了一个问题——AI预测药物活性,到底能不能用来做真实的药物设计决策?

答案比很多人想象的要复杂。
先说一个背景概念:什么是”预测药物活性”
做药的核心挑战之一,是搞清楚一个小分子药物和它的靶标蛋白结合得有多紧。结合得越紧,通常意味着药效越强(当然还有很多其他因素,但结合力是基础)。这个”有多紧”,在计算化学里用”结合自由能”来量化,单位是kcal/mol。
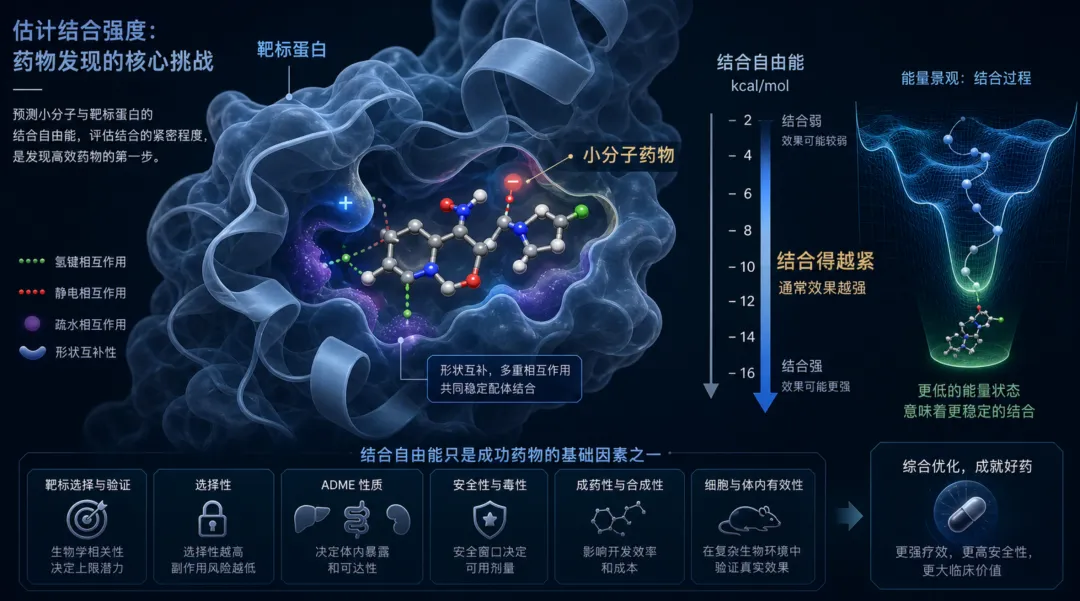
问题是,精确计算这个数字非常难。目前最靠谱的方法叫相对结合自由能计算(RBFE)——不直接算绝对值,而是算“分子A比分子B结合力强多少”。这正好对应药物设计中最常见的场景:你手上有一堆候选分子,需要排出哪个更好。
在这个领域,Schrödinger公司的FEP+是公认的业界标杆,精度高、稳定性好,但它是商业软件,价格不菲,代码也不开源。所以整个领域一直有两个平行的追问:开源物理方法能不能追上FEP+?AI方法能不能绕过物理计算直接给答案?
字节这篇文章同时回答了这两个问题。
第一个答案:开源物理方法终于接近FEP+了
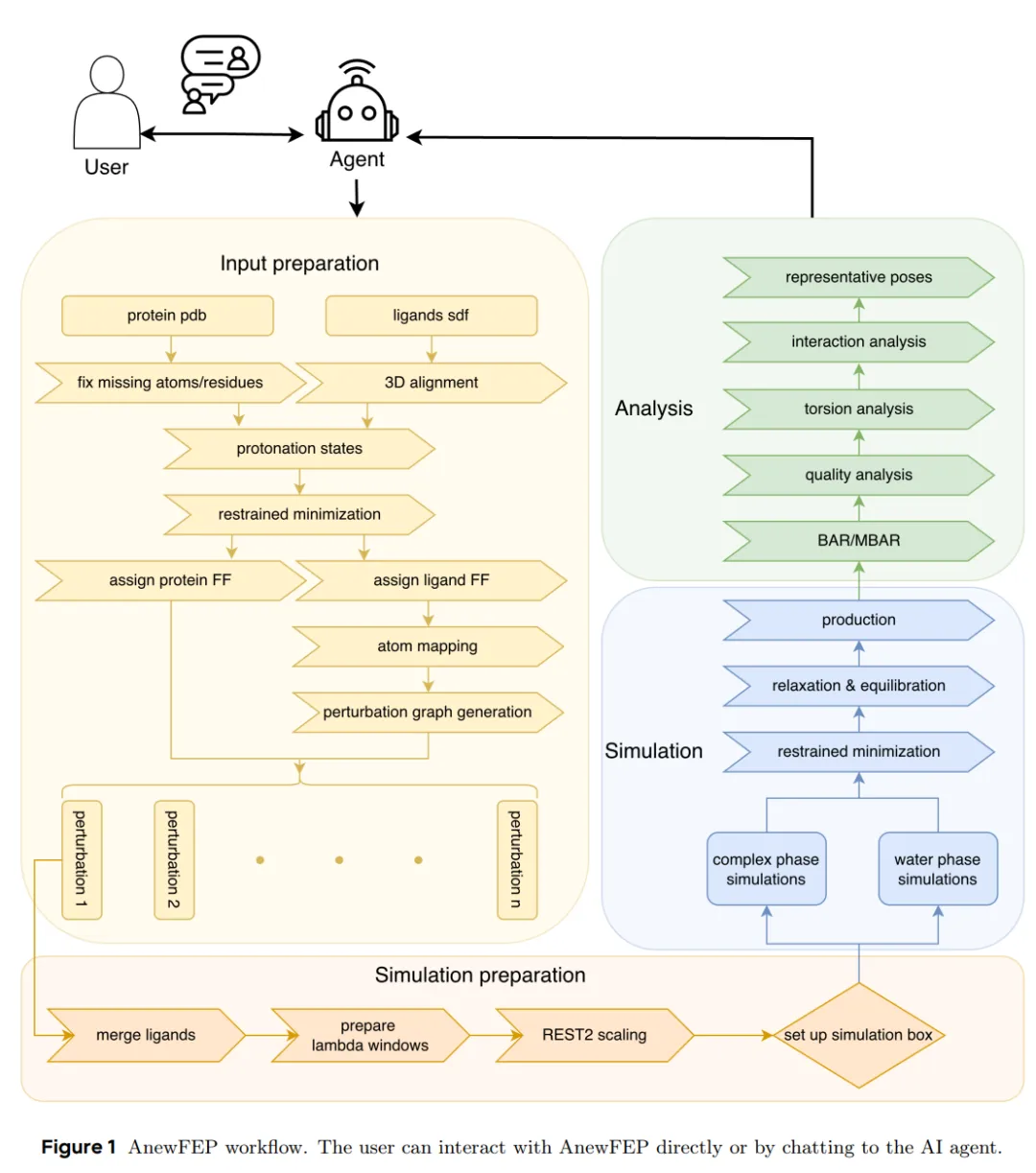
字节推出了AnewFEP,一个基于开源分子动力学软件GROMACS搭建的RBFE计算平台。它的核心包括一个自研的机器学习力场AnewFF(可以理解为用ML方法训练出来的一套描述原子间作用力的参数),加上REST2增强采样(一种帮助模拟更充分探索分子构象空间的技术),以及一系列针对已知失败模式的数值和协议改进。整个工作流从蛋白/配体输入到最终的自由能预测实现了端到端自动化,并集成了一个对话式AI agent作为交互界面——用户可以通过自然语言指令让agent自动设置计算任务、收集数据、分析结果,甚至执行复杂的分子筛选(比如”筛选出与靶标形成某个关键氢键且预测ΔG低于-9 kcal/mol的候选分子”)。底层的物理计算由传统MD引擎执行,agent负责任务编排和结果解读。
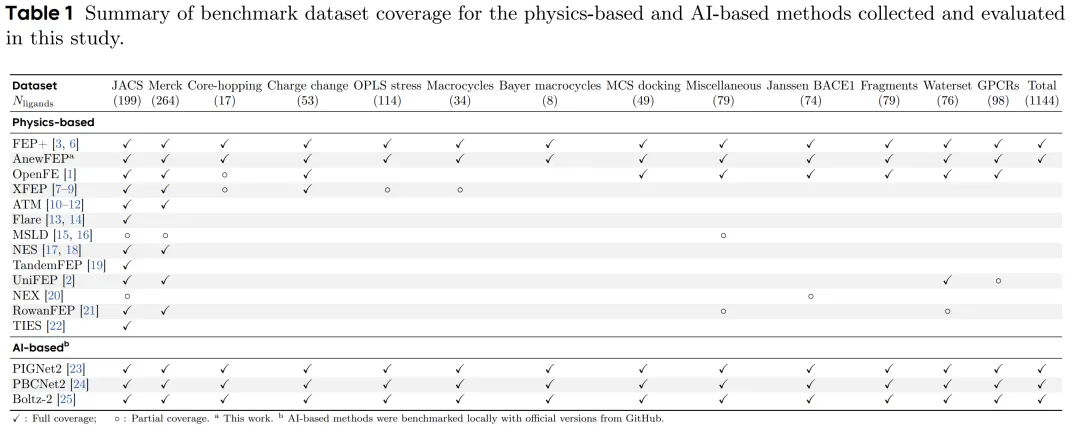
在一个包含1144个化合物、覆盖14种不同类型化学变换的公开测试集上,AnewFEP的整体精度达到了pairwise RMSE 1.44 kcal/mol。作为对比,FEP+在同一测试集上是1.25 kcal/mol。
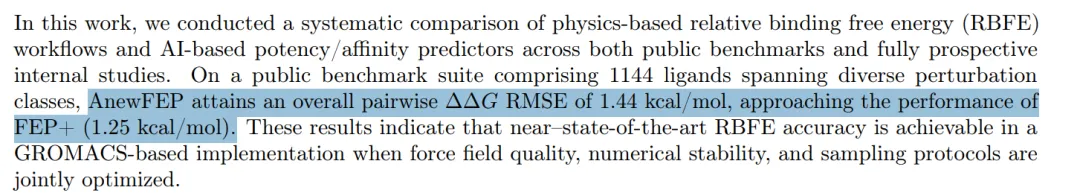
1.44 vs 1.25——看起来差距不大,但之前开源方案和FEP+的差距通常在0.5-0.7 kcal/mol甚至更大。比如在经典的Merck数据集上,FEP+是1.34,深势科技的UniFEP是1.82,Open Free Energy联盟的OpenFE是2.00。AnewFEP是第一个在全覆盖benchmark上真正逼近FEP+的非Schrödinger方案。

而且字节没有挑软柿子捏——core-hopping(大幅度骨架改造)、电荷变化、OPLS stress test(专门设计来暴露力场弱点的测试集)、大环化合物、GPCR靶标,这些公认难的类别全都跑了。
AnewFEP怎么做到的?论文里有四个非常扎实的case study,展示了具体的误差来源和修复方案。简单说,精度提升不是来自一个大的理论突破,而是来自一系列针对性的工程优化——修正特定原子的范德华半径参数、调整扭转势能面、控制蛋白骨架的构象漂移、重新设计炼金路径(alchemical pathway)的λ调度方案。每一个修复看起来都很”局部”,但累积起来就是从2.0降到1.44的差距。
第二个答案:AI在公开数据上很能打,但换到真实项目就不行了
这才是这篇文章最引发讨论的部分。字节在同一个公开benchmark上测了三个AI方法。其中表现最好的是Boltz-2,一个最近很火的蛋白-配体结构预测基础模型,它能同时预测蛋白和药物分子的三维结构,并输出一个亲和力估计。
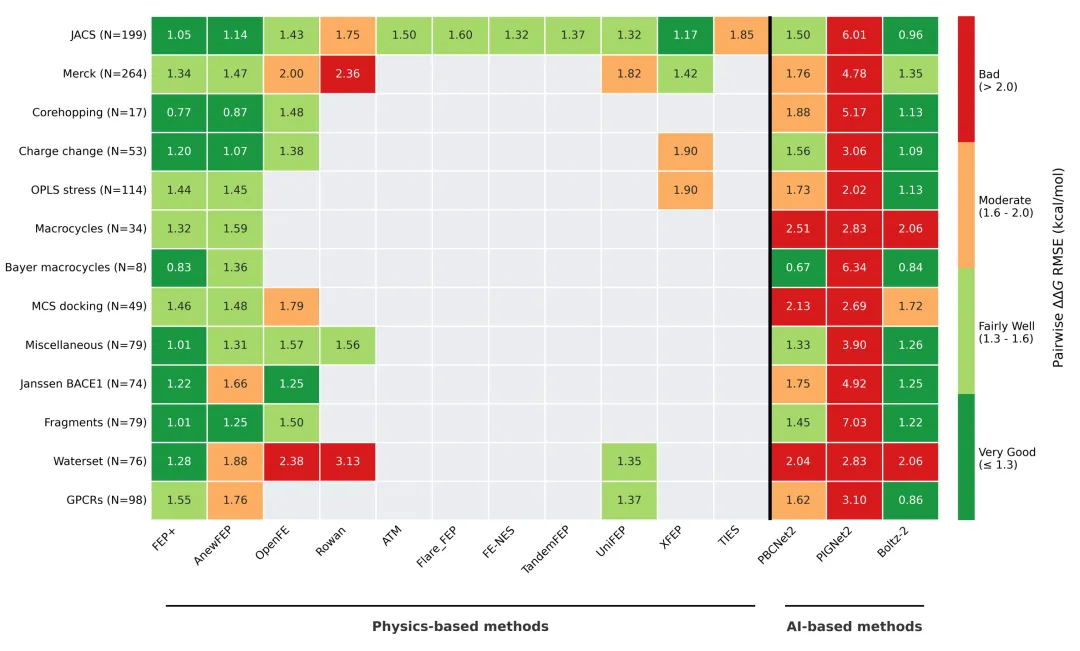
Boltz-2在公开数据上的表现相当亮眼:整体RMSE 1.25 kcal/mol,和FEP+打平。如果你只看这个数字,很容易得出”AI已经追上物理方法”的结论。
但字节做了一件大多数AI论文不会做的事:他们拿出了内部真实项目的数据来验证。这些数据全部来自他们自己从头设计的de novo分子——就是全新的、不在任何公开数据集中的化合物。
结果非常残酷。
在这个真实前瞻性数据集上,Boltz-2的R²掉到了0.01(满分1.0,越高越好),Spearman相关系数只有0.05(衡量排序能力,越接近1越好)。翻译成大白话:预测值和实验值之间几乎没有任何关联,排序能力约等于随机猜。
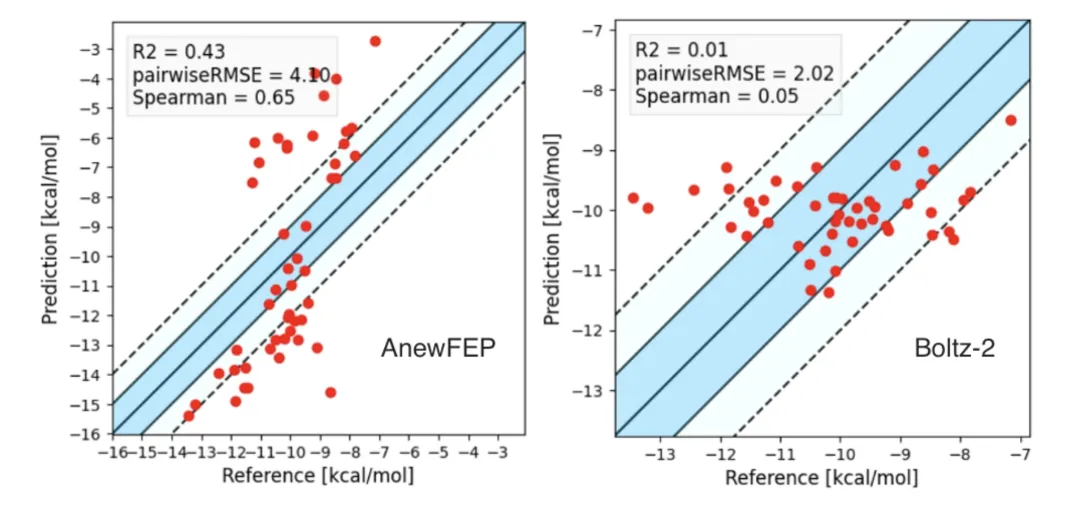
同样的数据,AnewFEP的R²是0.43,Spearman是0.65——虽然算不上完美,但至少还能有效区分哪些分子结合力强、哪些弱。在药物设计中,能排序就意味着能指导决策。
为什么AI在新分子上会崩?
论文里有一个特别有说服力的细节。在一个特定的蛋白体系中,结合口袋里有一个磷酸辅因子(可以理解为蛋白质工作时需要的一个”配件”)。对物理方法来说,辅因子在不在会显著改变结合口袋的电荷分布和空间环境,所以FEP计算的结果会因此产生很大差异——这在物理上完全合理。但Boltz-2无论加不加这个辅因子,预测结果几乎一样。

这说明Boltz-2大概率不是在”理解”分子间的物理化学相互作用,而是在从训练数据中学到的统计规律里做模式匹配。当你给它的分子长得像训练数据里见过的分子时,模式匹配是有效的,所以公开benchmark上成绩好看。但当你给它的是全新骨架、全新结合模式的分子时,它见都没见过类似的模式,预测就崩了。
这篇文章的真正价值
字节这篇文章的立场很值得玩味。他们自己就是AI团队,AnewFF本身就是机器学习力场,AnewFEP的工作流里甚至集成了一个对话式AI agent来辅助药物设计流程。他们一点都不反AI。
但他们用自己的内部数据,诚实地呈现了一个事实:AI方法在精心策划的公开benchmark上可以非常亮眼,但在前瞻性药物发现中——当化学空间、结合模式和靶标微环境都偏离训练分布时——AI的预测能力可能会大幅下降,而物理方法因为是从基本原理出发做计算,在这种情况下更加稳健。
论文最后的结论说得很务实:AI可以用于快速初筛,帮你从大量候选分子中快速缩小范围。但到了lead optimization阶段,需要对一小批精心设计的全新分子做精确排序时,物理方法依然是不可或缺的安全网。两者是互补关系,不是替代关系。
对整个AI制药领域来说,这篇文章提了一个重要的警醒:我们需要更好的评测范式。仅仅在公开benchmark上刷分,不足以证明一个模型在真实药物发现中有用。能不能在前瞻性的、分布外的化学空间上也报告性能?这可能比在已有benchmark上再提升0.1个百分点更有意义。Benchmark ≠ 实战。这句话,不管你做AI还是做药,都值得记住。

